
Abstract
The 3p21.3 RASSF1A tumour suppressor gene (TSG) provides a paradigm for TSGs inactivated by promoter methylation rather than somatic mutations. Recently, we identified frequent promoter methylation without somatic mutations of SLIT2 in lung and breast cancers, suggesting similarities between SLIT2 and RASSF1A TSGs. Epigenetic inactivation of RASSF1A was first described in lung and breast cancers and subsequently in a wide range of human cancers including neuroblastoma, Wilms' tumour and renal cell carcinoma (RCC). These findings prompted us to investigate SLIT2 methylation in these three human cancers. We analysed 49 neuroblastomas (NBs), 37 Wilms' tumours and 48 RCC, and detected SLIT2 promoter methylation in 29% of NB, 38% of Wilms' tumours and 25% of RCC. Previously, we had demonstrated frequent RASSF1A methylation in the same tumour series and frequent CASP8 methylation in the NB and Wilms' tumour samples. However, there was no significant association between SLIT2 promoter methylation and RASSF1A or CASP8 methylation in NB and RCC. In Wilms' tumour, there was a trend for a negative association between RASSF1A and SLIT2 methylation, although this did not reach statistical significance. No associations were detected between SLIT2 promoter methylation and specific clinicopathological features in the tumours analysed. These findings implicate SLIT2 promoter methylation in the pathogenesis of both paediatric and adult cancers and suggest that further investigations of SLIT2 in other tumour types should be pursued. However, epigenetic inactivation of SLIT2 is less frequent than RASSF1A in the tumour types analysed.
Similar content being viewed by others


Main
Methylation of CpG dinucleotides in the promoter regions of tumour suppressor genes (TSGs) producing transcriptional silencing is a major mechanism of TSG inactivation in many human cancers (Baylin et al, 1998; Jones, 1999; Tycko, 2000; Costello and Plass, 2001). The frequency of TSG inactivation by de novo methylation varies between TSGs and between cancers. Initially, epigenetic silencing of TSGs was reported as an alternative, but a minor mechanism of inactivation for TSGs such as RB1 and VHL (Greger et al, 1994; Herman et al, 1994). However, recently a new class of TSGs has been recognised for which epigenetic silencing is the overwhelming mechanism of inactivation and somatic mutations are rare. This paradigm is exemplified by the RASSF1A 3p21.3 TSG, which is methylated in a wide range of human cancers (Dammann et al, 2000; Agathanggelou et al, 2001; Astuti et al, 2001; Burbee et al, 2001; Dreijerink et al, 2001; Morrissey et al, 2001).
Recently, we identified frequent promoter region hypermethylation of SLIT2 in lung and breast cancers (Dallol et al, 2002a). SLIT2 maps to 4p15.2 and encodes a human orthologue of the Drosophila Slit protein, a secreted glycoprotein, which regulates axon guidance, branching and neural migration during development of the central nervous system (Brose et al, 1999). Slit exerts its effects as a diffusible chemorepellent via its receptor Roundabout (Robo) (Stein and Tessier-Lavigne, 2001). In mammals, three distinct slit genes (slit1, slit2, and slit3) and three distinct robo genes (robo1, robo2, rig-1) have been identified. In lung and breast cancers, SLIT2 mutations were not detected despite frequent promoter methylation (similar to RASSF1A) (Dallol et al, 2002a). Methylation of RASSF1A was first reported in lung and breast cancers, but subsequently we and others demonstrated RASSF1A methylation in renal cell carcinoma (RCC) and in common childhood tumours such as neuroblastoma (NB) and Wilms' tumour (Astuti et al, 2001; Dreijerink et al, 2001; Lo et al, 2001; Morrissey et al, 2001; Wagner et al, 2002). These findings prompted us to extend our studies of SLIT2 inactivation by studying SLIT2 promoter methylation in NB, Wilms' tumour and RCC.
Materials and methods
Patients and samples
A total of 134 tumour samples were analysed (49 NBs, 37 primary Wilms' tumours and 48 adult RCC). Details of the tumours have been published previously (Astuti et al, 2001; Wagner et al, 2002). DNA was extracted from tumour and normal tissues (blood or matched kidney) by standard methods.
Sodium bisulphite modification
Sodium bisulphite modification was carried out using an adapted method (Herman et al, 1996). Genomic DNA (0.5–1 μg) was denatured at 37°C for 10 min in 0.3 M NaOH. Unmethylated cytosines were sulphonated by incubation in 3.12 M sodium bisulphite/1 M hydroquinone (pH 5) at (95°C (30 s) 50°C (15 min)) × 20 cycles. The resulting sulphonated DNA was purified using the Wizard DNA clean-up system (Promega, Southampton, UK), according to the manufacturer's instructions, except that DNA was eluted with distilled water (50 μl) at room temperature. Following elution, DNA was desulphonated in 0.3 M NaOH for 5 min at room temperature, then the DNA was precipitated with NaOAc (5 μl of 3 M) and ethanol (125 μl of 100%) overnight at −20°C and resuspended in 50 μl distilled water.
Methylation-specific polymerase chain reaction (PCR)
The CpG island within the putative SLIT2 promoter has been described in detail previously (Dallol et al, 2002a). Methylation Specific PCR (MSP) analysis was performed essentially using primers and conditions as described in Dallol et al, 2002a). Combined Bisulphite Restriction Analysis (CoBRA) analysis was carried out as follows. In brief, the putative promoter region from nt −761 to −212 (relative to the translation start site) was amplified with primers reported previously (Slit2MODF-(5′-GGGAGGTGGGATTGTTTAGATATTT-3′ and Slit2MODR2 (5′-CAAAAACTCCTTAAACAACTTTAAATCCTAAAA-3′). From this reaction, one out of 50 of the volume was used as a template for a nested PCR with primers Slit2MODF as above and Slit2MODR (5′-ACTAAAACTTCCAACAACTACTAAAATACAAAAA-3′) to produce a 418 bp product (PCR conditions: 95°C for 10 min, followed by 30–40 cycles of 1 min denaturation at 95°C, 1 min annealing at 54°C, and 2 min extension at 74°C). PCR products were then digested by BstU1 restriction enzyme to assess the methylation status of samples.
Sequencing of PCR products
MSP and CoBRA products were excised from agarose gels and extracted using the QIAquick Gel Extraction Kit (Qiagen, Crawley, UK, West Sussex), according to the manufacturer's instructions. Products were confirmed by direct sequencing from the forward PCR primer using ABI Prism® BigDye™Terminators Cycle Sequencing Kit according to the manufacturer's instructions and run using ABI 377 automatic sequencers.
5-aza-2′-deoxycytidine treatment of cell lines
5-aza-2′-deoxycytidine (5-aza-dC, Sigma, Poole, UK) was freshly prepared in ddH2O (at 2 mg ml−1) and filter-sterilised. A total of 0.5–1 × 106 cells were plated in 75 cm2 flask in RPMI 1640 media supplemented with 10% foetal calf serum and left to settle for 24 h (day 0). Kidney and NB cell lines were treated with 2 μ M 5-aza-dC for a total of 5 days. Kidney and NB have a different rate of growth. Kidney tumour cell lines were treated with 5-aza-dC on days 1, 3 and 5 and harvested on day 6, with a medium change in days 2 and 4 (Nojima et al, 2001). Neuroblastoma cell lines were treated on days 1 and 4. The medium was changed 24 h after treatment and then every 3 days. RNA was prepared after treatment using the RNeasy kit (Qiagen) according the manufacturer's guidelines. SLIT2 gene expression was ascertained by RT–PCR using the primers 5′-GGTGTCCTCTGTGATGAAGAG-3′ and 5′-GTGTTTAGGAGACACACCTCG-3′, resulting in a product size 387 bp. As a control, the GAPDH primers used were: 5′-GACCCCTTCATGACCTCAACTACA-3′ and 5′-CTAAGCAGTTGGTGGTGCAGGA-3′, resulting in a PCR product of 369 bp.
Microsatellite repeat analysis – loss of heterozygosity
By searching GDB and the UCSC assembly of the human genome sequence, we identified D4S1546 as the closest marker to SLIT2 (within 100 kb). A 4p15.2 allele loss was assessed with the D4S1546 marker. (PCR conditions: 95°C for 5 min followed by 35 cycles of 95°C for 30 s, 52°C (55°C) for 30 s, and 72°C for 30 s and a final extension of 10 min at 72°C).
Statistical analysis
Fisher's exact test was used as appropriate. P-values of <0.05 were considered to be statistically significant.
Results
SLIT2 methylation status in NB
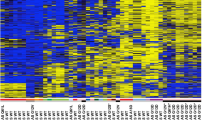
SLIT2 promoter methylation status was analysed in 49 primary NB tumours and 29% (14 out of 49) demonstrated SLIT2 CpG island promoter methylation (Figure 1A). Promoter CpG island methylation was confirmed by direct sequencing of five clones from one tumour (Figure 4B). We also analysed eight NB cell lines for SLIT2 methylation by restriction digestion and two cell lines (SK-N-F1 and SK-N-SH) were found to be partially methylated. SLIT2 methylation was detected in one out of 49 corresponding normal blood samples.

Methylation analysis of SLIT2 by MSP in neuroblastoma tumours (A) and Wilm's tumours (B) and by CoBRA in RCC cell lines and primary kidney tumours and corresponding normal tissue (C). (A and B). Bisulphite-modified DNA was amplified with primers specific for unmethylated (U) and methylated (M) DNA. Tumours positive for SLIT2 methylation are depicted by the presence of a 160 bp product when using specific primers for methylated DNA. N=blood; T=tumour tissue. (C) Bisulphite-modified DNA was amplified by nested PCR and then digested with BstU1 restriction enzyme for 4 h at 60°C, uncut (Un) and cut (C). SKRC 18 is partially methylated while SKRC 47 is unmethylated. The RCC tumour shown above is completely methylated.

SLIT2 methylation profile. Illustration of the SLIT2 methylation pattern detected in (A) kidney tumour cell lines (SKRC 39, SKRC 18 and SKRC 47) and (B) neuroblastoma tumours (NB107) and Wilm's tumours (WT244). The CpG island numbered according to Dallol et al (2002a).
To investigate the 4p15.2 allelic status of NBs with SLIT2 methylation, we typed 13 methylated tumours for loss of heterozygosity (LOH) at D4S1546 that maps close to SLIT2. In all, 33% (four out of 11) of informative tumours demonstrated allele loss consistent with homozygous SLIT2 inactivation (Figure 2).

Genotyping of marker D4S1546 in neuroblastoma tumours. N and T, matched DNA samples from blood (N) and tumour tissue (T). Tumours 125 and 162 showed LOH, while tumour 133 shows retention of allele. The position of the lost allele is indicated by the arrows.
SLIT2 promoter methylation is associated with transcriptional silencing
To determine the relationship of SLIT2 promoter region CpG island methylation and SLIT2 transcript expression in the NB cell lines SK-N-F1 and SK-N-SH, we treated the cells with the demethylating agent, 5-aza-dC, for 5 days. The 5-aza-dC treatment significantly increased SLIT2 expression in both cell lines, but there was little or no change in the expression of GAPDH expression after the 5-aza-dC treatment (Figure 3).

SLIT2 expression by RT–PCR in neuroblastoma (SK-N-SH) and renal cell carcinoma cell lines (SKRC 39, SKRC 18 and SKRC 47), without (−) and with (+) 5-aza-2′-deoxycytidine (5-aza-dc) treatment. Cells were treated for up to 5 days with 5-aza-dc and expression was analysed by RT–PCR. The methylated cell lines (SK-N-SH, SKRC 39, SKRC 18) show an increase in SLIT2 expression after 5-aza-dc treatment, while the unmethylated cell line SKRC 47 presents no changes in SLIT2 expression before and after 5-aza-dc treatment.
SLIT2, CASP8 and RASSF1A methylation status in NB
Previously, we reported that RASSF1A and caspase 8 (CASP8) promoter methylation occurred in 55 and 40% of NBs, respectively. To determine whether there was any relationship between SLIT2 promoter methylation and de novo methylation of RASSF1A and CASP8 promoters, we compared the frequencies of RASSF1A and CASP8 methylation in tumours with and without SLIT2 methylation (using previously published RASSF1A and CASP8 methylation data (Astuti et al, 2001). CASP8 methylation was detected in 36% of SLIT2 methylated and 41% SLIT2 unmethylated tumours (P=1.0). RASSF1A promoter methylation was more frequent in tumours with SLIT2 promoter methylation (77 vs 59%), but this did not reach statistical significance (P=0.32).
Neuroblastoma clinicopathological features and SLIT2 methylation status
We compared the results of SLIT2 methylation status in our tumour series to the previously reported results for allelic loss of 1p or 3p loss, N-myc amplification and 17q gain (Martinsson et al, 1997). There was no correlation between SLIT2 methylation and 1p allele loss (22% of SLIT2 methylated and 22% of unmethylated tumours, P=1.0), 3p allele loss (25 vs 12%, P=0.57), 17q gain (50 vs 65%, P=0.62) or N-myc amplification status (15 vs 13%, P=1.0). There was no association between tumour stage and SLIT2 methylation status: SLIT2 methylation was present in 33% of stage 1, 2 and 4S tumours and in 26% of stage 3 and 4 tumours (P=0.73).
Methylation analysis of SLIT2 in primary Wilms' tumours
Next, we proceeded to analyse SLIT2 promoter methylation status in 37 Wilms' tumours that had been investigated previously for RASSF1A and CASP8 promoter methylation status (Wagner et al, 2002 and unpublished observations). In total, 38% (14 out of 37) Wilms' tumours demonstrated SLIT2 CpG island promoter methylation (Figure 1B). Promoter CpG island methylation was confirmed by direct sequencing in one tumour (Figure 4B). All SLIT2 methylated tumours contained unmethylated SLIT2 alleles that might be attributable to the presence of contaminating normal tissue (tumour samples were not microdissected). SLIT2 methylation was detected in zero of six normal tissue samples corresponding to the methylated tumours.
To investigate the 4p15.2 allelic status of Wilms' tumours with SLIT2 methylation, we typed six methylated tumours for LOH at D4S1546. None of three informative tumours demonstrated D4S1546 allele loss.
Methylation of SLIT2 and other cancer genes in primary Wilms' tumours
To investigate the relationships between SLIT2 promoter methylation and de novo methylation of RASSF1A and CASP8, we compared the frequencies of CASP8 and RASSF1A methylation in tumours with and without SLIT2 methylation. In tumours with SLIT2 methylation, CASP8 and RASSF1A were methylated in 43% (six out of 14) and 36% (five out of 14), respectively. In tumours without SLIT2 methylation, CASP8 and RASSF1A promoter methylation was detected in 39% (nine out of 23) and 70% (16 out of 23), respectively. Thus, although there was no association between SLIT2 and CASP8 methylation, there was an inverse relationship between SLIT2 and RASSF1A methylation, although this did not reach statistical significance (P=0.09).
SLIT methylation status and clinicopathological status
The frequency of relapse in Wilms' tumours with SLIT2 methylation was similar to that without SLIT2 methylation (21% (three out of 14) and 17% (four out of 23), respectively), and there was no significant association between SLIT2 methylation and advanced stage tumours (the frequency of stage 3 and 4 tumours in the SLIT2 methylated and unmethylated groups was 45% (five out of 11) and 63% (12 out of 19), respectively).
Methylation analysis of SLIT2 in primary RCC
We detected SLIT2 promoter methylation in 25% (12 out of 48) primary RCC and in 75% (six out of eight) RCC cell lines (Figure 1C). Promoter CpG island methylation was confirmed by direct sequencing of five clones from RCC cell lines and 2 tumours (Figure 4A). SLIT2 promoter methylation was also detected in one out of 12 of the matching normal kidney tissue samples for methylated tumours. All RCC with SLIT2 methylation also contained unmethylated SLIT2 alleles, which might be attributable to the presence of contaminating normal tissue (tumour samples were not microdissected). Loss of heterozygosity at D4S1546 was not detected in 10 informative RCC with SLIT2 methylation.
SLIT2 promoter methylation is associated with transcriptional silencing in an RCC cell line
We investigated the possible association between the SLIT2 promoter region CpG island methylation and SLIT2 transcript expression in a panel of RCC cell lines (SKRC 18, SKRC 39, SKRC 45, SKRC 47, SKRC 54, KTCL 26,UMRC-2 and 786-0). Cells were treated with the demethylating agent 5-aza-dC for 5 days. Except for SKRC 45 and SKRC 47 (both unmethylated for SLIT2), SLIT2 expression was significantly increased in the kidney tumour cell lines after 5-aza-dC treatment. GAPDH expression levels were equal in both 5-aza-dC-treated and untreated cell lines (Figure 3).
Methylation status of SLIT2 and inactivation of RASSF1A and VHL in primary RCC
Previously, we analysed primary RCC for RASSF1A methylation and inactivation of the VHL tumour suppressor gene. There was no association between SLIT2 methylation and the presence of VHL mutation in clear cell RCC, and the frequency of RASSF1A methylation was similar in RCC with and without SLIT2 methylation (25 and 21%, respectively).
SLIT2 methylation status and clinicopathological status
The frequency of SLIT2 methylation in clear cell RCC (24%, nine out of 37) was similar to that found in all tumour types. There was no significant association between SLIT2 methylation status and grade or TNM status.
Discussion
Previously, we (a) identified SLIT2 promoter methylation in 53% non-small-cell lung cancer, 36% small-cell lung cancer and 43% of breast cancers, (b) demonstrated that promoter methylation is associated with reversible transcriptional silencing and (c) determined that restoration of SLIT2 expression suppressed tumour growth in in vitro studies (Dallol et al, 2002a). Thus having established SLIT2 as a lung and breast cancer suppressor gene, we have now identified frequent SLIT2 hypermethylation in paediatric cancers and in RCC and, recently, in 59% of gliomas (Dallol et al, 2003). Slit protein binds to the roundabout receptor and acts as a midline repellent to guide axonal development during embryogenesis (Rajagopalan et al, 2000; Simpson et al, 2000; Wong et al, 2001) In humans, three SLIT orthologues have been identified, but to date only SLIT2 has been implicated in cancer. In mice, Robo1 inactivation produces delayed lung maturation and bronchial hyperplasia (Xian et al, 2001) and we have demonstrated de novo ROBO1 promoter methylation in 19% primary invasive breast carcinomas and 18% primary clear cell RCC, although somatic mutations were not identified (Dallol et al, 2002b).
The characterisation of tumour-specific TSG methylation profiles provides insights into the molecular pathology of the tumour and can be used to establish tumour-specific methylation profiles (Esteller et al, 2001a). It is anticipated that such profiles will be useful for diagnosis and clinicopathological classification. In colorectal cancer, the identification of a subset of tumours with widespread de novo methylation of TSG and mismatch repair genes (‘the methylator phenotype’) (Toyota et al, 1999) has been of considerable interest and it is unclear as to whether similar subtypes will be a feature of other cancers.
The molecular pathology of sporadic NB has been investigated extensively by molecular cytogenetic and LOH analysis. Frequent alterations include N-myc amplification (20–25%) and gain of genetic material at 17q23–qter (∼50% of tumours). Neuroblastoma TSGs have been mapped by LOH studies to 1p36 (∼35% in primary tumours show LOH), 11q23 (44%), 14q23–qter (22%) and 3p (15%) (Ejeskar et al, 1998; Maris and Matthay, 1999). However, specific gene mutations have not been defined, although we did identify RASSF1A methylation in 55% of NBs and that CASP8 methylation occurs in ∼50% of tumours (Teitz et al, 2000; Astuti et al, 2001). Caron et al (1996) reported 4p allele loss in ∼20% of NB and we have now identified promoter methylation of the 4p15.2 candidate TSG SLIT2 in 29% of NBs. Thus, epigenetic inactivation of SLIT2 is a common feature of NB, although less frequent than methylation of CASP8 and RASSF1A. Harada et al (2002b) reported that NB patients with CASP8 methylation were older than those without tumour methylation, but to date, CASP8, RASSF1A or SLIT2 methylation has not been associated with specific clinicopathological, cytogenetic or molecular features of NB. However, very large studies may be needed to identify significant prognostic correlations in the presence of a large number of potential variables. We did not find clear evidence of a ‘methylator phenotype’, although methylation of RASSF1A was more common in tumours with SLIT2 methylation than in those without SLIT2 methylation and Harada et al (2002b) reported an association between RASSF1A and CASP8 methylation in NB tumours. However, we found no association between SLIT2 and CASP8 methylation. The frequency of SLIT2 methylation in NB was less than that for RASSF1A and CASP8, but is still a significant finding as other candidate TSGs that demonstrate frequent promoter methylation in some cancers (e.g. p16INK4A, MGMT, RARâ, DAPK, APC, GSTP1, CDH1 and CDH13) are rarely methylated in NB (Harada et al, 2002a).
Although de novo methylation and silencing of H19 in Wilms' tumours was first reported some years ago (Steenman et al, 1994; Taniguchi et al, 1995), epigenetic changes have not been investigated in great detail in Wilms' tumour. Recently, we reported frequent CASP8 (43%) and RASSF1A (56%) promoter methylation in Wilms' tumours and the present study has demonstrated that SLIT2 methylation represents a further frequent epigenetic change in Wilms' tumours. To date, we have not identified an association between CASP8, RASSF1A and SLIT2 methylation in individual tumours, so there is little evidence of a methylator phenotype in a subset of Wilms' tumours. Indeed there was a negative, albeit statistically insignificant, correlation between RASSF1A and SLIT2 methylation. This finding merits further investigation as it could indicate that the simultaneous inactivation of specific TSGs might be disadvantageous in specific cancer types. Although p16INK4a(CDKN2a) promoter methylation has been reported in advanced stage Wilms' tumours (Arcellana-Panlilio et al, 2000), this trend was not significant in our series, and to date no specific clinicopathological features have been associated with SLIT2, CASP8 and RASSF1A methylation. In our Wilms' tumour series, the frequency of SLIT2 promoter methylation in Wilms' tumours was similar to that for CASP8 but higher than that for TSGs, which may show frequent promoter methylation in other tumour types, for example, MGMT (30%), NORE1A (15%), p14ARF (15%), p16INK4a (10%), DAPK (11%), CRBP1 (9%), RARB2 (0%), CDH13 (0%) and CDH1 (3%) (Morris et al, 2003).
Renal cell carcinoma is the most common adult tumour and the majority (∼75%) of RCC are classified as clear cell RCC, with papillary being the most frequent nonclear cell histopathological subtype (∼15% of all cases) (Storkel and van den Berg, 1995). The most frequent genetic change in RCC is somatic inactivation of the VHL TSG (usually be mutation and loss, but promoter methylation may also occur), although VHL inactivation is specific for clear cell RCC (Foster et al, 1994; Gnarra et al, 1994; Clifford et al, 1998). Combining the results of the current study with previous investigations, frequent epigenetic changes (⩾20%) in RCC include promoter methylation of RASSF1A, TIMP3, DAPK, SLIT2, MT1G and GSTP1 (Esteller et al, 2001a; Nojima et al, 2001, Dreijerink et al, 2001; Morrissey et al, 2001, Morris et al, 2003). In contrast, promoter methylation not at (or rarely at) SDHB, RARB2, p16INK4a and CDH13 is uncommon (<5%). To date, apart from VHL, none of the epigenetic changes in RCC have been associated with specific clinicopathological features.
The failure to detect an association between clinicopathological stage and SLIT2 methylation status could indicate that SLIT2 methylation is an early event in tumorigenesis. In tumours such as colorectal cancer, where there is a well-validated adenoma–carcinoma sequence, it is possible to define the genetic changes associated with different stages of tumorigenesis. However, in sporadic cases of NB, Wilms' tumour and RCC, there is generally no well-defined pathway from precursor lesion to tumour (although nephroblastomatosis may be present in patients with Beckwith–Widemann syndrome and ‘early lesion RCC’ has been described in von Hippel-Lindau disease). Hence, we are unable to precisely define when SLIT methylation occurs in the pathogenesis of these tumours. However, it is known that TSG inactivation may be an early event in tumorigenesis. Thus, methylation may be the ‘second hit’ in familial cancer syndrome tumours (Prowse et al, 1997, Esteller et al, 2001b). Furthermore, in sporadic and familial adenomatous polyposis coli, TSG CpG island methylation may be detected in the earliest precursor lesion in colorectal carcinogenesis, aberrant crypt foci (Chan et al, 2002). Similarly, Zöchbauer-Müller et al (2001) suggested that TSG methylation may be a preneoplastic change in non-small-cell lung cancer. We have analysed previously RASSF1A promoter methylation status in normal, ductal-carcinoima-in situ (DCIS) and breast cancer trios. RASSF1A promoter hypermethylation was detected in 65% of invasive cancers and in 42% of corresponding DCIS but in none of the normal breast samples (Honorio et al, 2003). In all, 30% of DCIS without invasive breast cancer also underwent RASSF1A promoter hypermethylation, suggesting that inactivation of RASSF1A by CpG island methylation is an early event in breast tumorigenesis. Preliminary unpublished data also reveal SLIT2 methylation in DCIS samples (RE Dickinson and F Latif, unpublished). Thus, there is evidence that SLIT2 hypermethylation can be implicated in early tumorigenesis.
In breast and lung cancers, TSG promoter methylation SLIT2 appears to resemble TSGs such as RASSF1A, as epigenetic inactivation is more frequent than somatic mutations. RASSF1A methylation has been reported in a wide range of human cancers. We have demonstrated that SLIT2 methylation is common in paediatric and adult cancers, and further analysis of additional tumour types seems indicated. Frequent 4p allele loss has been reported in cancers that demonstrate SLIT2 methylation such as lung, breast and NB, and also in cancers in which SLIT2 methylation status has not been investigated including colorectal, bladder and head and neck cancers (Knowles et al, 1994; Rosin et al, 1995; Gryfe et al, 1997; Pershouse et al, 1997; Yustein et al, 1999; Girard et al, 2000). During development, the SLIT2 protein functions as a secreted chemorepellent so that restoration of SLIT2 function by reversal of epigenetic inactivation or administration of SLIT2 agonists might provide novel therapeutic opportunities for human cancers.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Agathanggelou A, Honorio S, Macartney DP, Martinez A, Dallol A, Rader J, Fullwood P, Chauhan A, Walker R, Shaw JA, Hosoe S, Lerman MI, Minna JD, Maher ER, Latif F (2001) Methylation associated inactivation of RASSF1A from region 3p21.3 in lung, breast and ovarian tumours. Oncogene 20: 1509–1518
Arcellana-Panlilio MY, Egeler RM, Ujack E, Pinto A, Demetrick DJ, Robbins SM, Coppes MJ (2000) Decreased expression of the INK4 family of cyclin-dependent kinase inhibitors in Wilms tumor. Genes Chromosomes Cancer 29: 63–69
Astuti D, Agathanggelou A, Honorio S, Dallol A, Martinsson T, Kogner P, Cummins C, Neumann HPH, Voutilainen R, Dahia P, Eng C, Maher ER, Latif F (2001) RASSF1A promoter region CpG island hypermethylation in phaeochromocytomas and neuroblastoma tumours. Oncogene 20: 7573–7577
Baylin SB, Herman JG, Graff JR, Vertino PM, Issa JP (1998) Alterations in DNA methylation: a fundamental aspect of neoplasia. Adv Cancer Res 72: 141–196
Brose K, Bland KS, Wang KH, Arnott D, Henzel W, Goodman CS, Tessier-Lavigne M, Kidd T (1999) Slit proteins bind Robo receptors and have an evolutionarily conserved role in repulsive axon guidance. Cell 96: 795–806
Burbee DG, Forgacs E, Zöchbauer-Müller S, Shivakumar L, Gao B, Randle D, Virmani A, Bader S, Sekido Y, Latif F, Fong K, Gazdar AF, Lerman MI, White M, Minna JD (2001) Epigenetic inactivation of RASSF1A in lung and breast cancers and malignant phenotype suppression. J Natl Cancer Inst 93: 691–699
Caron H, van Sluis P, Buschman R, Pereira do Tanque R, Maes P, Beks L, de Kraker J, Voute PA, Vergnaud G, Westerveld A, Slater R, Versteeg R (1996) Allelic loss of the short arm of chromosome 4 in neuroblastoma suggests a novel tumour suppressor gene locus. Hum Genet 97: 834–837
Chan AO, Broaddus RR, Houlihan PS, Issa JP, Hamilton SR, Rashid A (2002) CpG island methylation in aberrant crypt foci of the colorectum. Am J Pathol 160: 1823–1830
Clifford SC, Prowse AH, Affara NA, Buys CHCM, Maher ER (1998) Inactivation of the von Hippel-Lindau (VHL) tumour suppressor gene and allelic losses at chromosome arm 3p in primary renal cell carcinoma: evidence for a VHL-independent pathway in clear cell renal tumorigenesis. Genes Chromosomes Cancer 22: 200–209
Costello JF, Plass C (2001) Methylation matters. J Med Genet 38: 285–303
Dallol A, da Silva NF, Viacava P, Minna JD, Maher ER, Latif F (2002a) SLIT2, a human homologue of the Drosophila Slit2 gene, has tumor suppressor activity and is frequently inactivated in lung and breast cancers. Cancer Res 62: 5874–5880
Dallol A, Krex D, Hesson L, Eng C, Maher ER, Latif F (2003) Frequent epigenetic inactivation of the SLIT2 gene in gliomas. Oncogene 22: 4611–4616
Dallol A, Forgacs E, Martinez A, Sekido Y, Walker R, Kishida T, Rabbitts P, Maher ER, Latif F (2002b) Tumour specific promoter region methylation of the human homologue of the Drosophila Roundabout gene DUTT1 (ROBO1) in human cancers. Oncogene 21: 3020–3028
Dammann R, Li C, Yoon JH, Chin PL, Bates S, Pfeifer GP (2000) Epigenetic inactivation of a RAS association domain family protein from the lung tumour suppressor locus 3p21.3. Nat Genet 25: 315–319
Dreijerink K, Braga E, Kuzmin I, Geil L, Duh FM, Angeloni D, Zbar B, Lerman MI, Stanbridge EJ, Minna JD, Protopopov A, Li JF, Kashuba V, Klein G, Zabarovsky ER (2001) The candidate tumor suppressor gene, RASSF1A, from human chromosome 3p21.3 is involved in kidney tumorigenesis. Proc Natl Acad Sci USA 98: 7504–7509
Ejeskar K, Aburatani H, Abrahamsson J, Kogner P, Martinsson T (1998) Loss of heterozygosity of 3p markers in neuroblastoma tumours implicate a tumour-suppressor locus distal to the FHIT gene. Br J Cancer 77: 1787–1791
Esteller M, Corn PG, Baylin SB, Herman JG (2001a) A gene hypermethylation profile of human cancer. Cancer Res 61: 3225–3229
Esteller M, Fraga MF, Guo M, Garcia-Foncillas J, Hedenfalk I, Godwin AK, Trojan J, Vaurs-Barriere C, Bignon YJ, Ramus S, Benitez J, Caldes T, Akiyama Y, Yuasa Y, Launonen V, Canal MJ, Rodriguez R, Capella G, Peinado MA, Borg A, Aaltonen LA, Ponder BA, Baylin SB, Herman JG (2001b) DNA methylation patterns in hereditary human cancers mimic sporadic tumorigenesis. Hum Mol Genet 10: 3001–3007
Foster K, Prowse A, van den Berg A, Fleming S, Hulsbeek MMF, Crossey PA, Richards FM, Cairns P, Affara NA, Ferguson-Smith MA, Buys CHCM, Maher ER (1994) Somatic mutations of the von Hippel-Lindau disease tumour suppressor gene in non-familial clear cell renal carcinoma. Hum Mol Genet 3: 2169–2173
Girard L, Zöchbauer-Müller S, Virmani AK, Gazdar AF, Minna JD (2000) Genome-wide allelotyping of lung cancer identifies new regions of allelic loss, differences between small cell lung cancer and non-small cell lung cancer, and loci clustering. Cancer Res 60: 4894–4906
Gnarra JR, Tory K, Weng Y, Schmidt L, Wei MH, Li H, Latif F, Liu S, Chen F, Duh FM, Lubensky I, Duan DR, Florence C, Pozatti R, Walther MM, Bander NH, Grossman HB, Brauch H, Pomer S, Brooks JD, Isaacs WB, Lerman MI, Zbar B, Linehan WM (1994) Mutations of the VHL tumour suppressor gene in renal carcinoma. Nat Genet 7: 85–90
Greger V, Debus N, Lohmann D, Hopping W, Passarge E, Horsthemke B (1994) Frequency and parental origin of hypermethylated RB1 alleles in retinoblastoma. Hum Genet 94: 491–496
Gryfe R, Swallow C, Bapat B, Redston M, Gallinger S, Couture J (1997) Molecular biology of colorectal cancer. Curr Probl Cancer 21: 233–300
Harada K, Toyooka S, Maitra A, Maruyama R, Toyooka KO, Timmons CF, Tomlinson GE, Mastrangelo D, Hay RJ, Minna JD, Gazdar AF (2002a) Aberrant promoter methylation and silencing of the RASSF1A gene in pediatric tumors and cell lines. Oncogene 21: 4345–4349
Harada K, Toyooka S, Shivapurkar N, Maitra A, Reddy JL, Matta H, Miyajima K, Timmons CF, Tomlinson GE, Mastrangelo D, Hay RJ, Chaudhary PM, Gazdar AF (2002b) Deregulation of caspase 8 and 10 expression in pediatric tumors and cell lines. Cancer Res 62: 5897–5901
Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB (1996) Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA 93: 9821–9826
Herman JG, Latif F, Weng Y, Lerman MI, Zbar B, Liu S, Samid D, Duan DS, Gnarra JR, Linehan WM (1994) Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proc Natl Acad Sci USA 91: 9700–9704
Honorio S, Agathanggelou A, Schuermann M, Pankow W, Viacava P, Maher ER, Latif F (2003) Detection of RASSF1A aberrant promoter hypermethylation in sputum from chronic smokers and ductal carcinoma in situ from breast cancer patients. Oncogene 22: 147–150
Jones PA (1999) The DNA methylation paradox. Trends Genet 15: 34–37
Knowles MA, Elder PA, Williamson M, Cairns JP, Shaw ME, Law MG (1994) Allelotype of human bladder cancer. Cancer Res 54: 531–538
Lo KW, Kwong J, Hui ABY, Chan SYY, To KF, Chan SC, Chow LSN, Teo PML, Johnson PJ, Huang DP (2001) High frequency of promoter hypermethylation of RASSF1A in nasopharyngeal carcinoma. Cancer Res 61: 3877–3881
Maris JM, Matthay KK (1999) Molecular biology of neuroblastoma. J Clin Oncol 17: 2264–2279
Martinsson T, Sjoberg RM, Hallstensson K, Nordling M, Hedborg F, Kogner P (1997) Delimitation of a critical tumour suppressor region at distal 1p in neuroblastoma tumours. Eur J Cancer 33: 1997–2001
Morris MR, Hesson LB, Wagner KJ, Morgan NV, Astuti D, Lees RD, Cooper WN, Lee JA, Gentle D, Macdonald F, Kishida T, Grundy R, Yao M, Latif F, Maher ER (2003) Multigene methylation analysis of Wilms' tumour and adult renal cell carcinoma. Oncogene 22: 6794–6801
Morrissey C, Martinez A, Zatyka M, Agathanggelou A, Honorio S, Astuti D, Morgan NV, Moch H, Richards FM, Kishida T, Yao M, Schraml P, Latif F, Maher ER (2001) Epigenetic inactivation of the RASSF1A 3p21.3 tumor suppressor gene in both clear cell and papillary renal cell carcinoma. Cancer Res 61: 7277–7281
Nojima D, Nakajima K, Li LC, Franks J, Ribeiro L, Ishii N, Dahiya R (2001) CpG methylation of promoter region inactivates E-cadherin gene in renal cell carcinoma. Mol Carcinogen 32: 19–27
Pershouse MA, El-Naggar AK, Hurr K, Lin H, Yung WK, Steck PA (1997) Deletion mapping of chromosome 4 in head and neck squamous cell carcinoma. Oncogene 14: 369–373
Prowse AH, Webster AR, Richards FM, Richard S, Olschwang S, Resche F, Affara NA, Maher ER (1997) Somatic inactivation of the VHL gene in von Hippel-Lindau disease tumors. Am J Hum Genet 60: 765–771
Rajagopalan S, Vivancos V, Nicolas E, Dickson BJ (2000) Selecting a longitudinal pathway: Robo receptors specify the lateral position of axons in the Drosophila CNS. Cell 103: 1033–1045
Rosin MP, Cairns P, Epstein JI, Schoenberg MP, Sidransky D (1995) Partial allelotype of carcinoma in situ of the human bladder. Cancer Res 55: 5213–5216
Simpson JH, Bland KS, Fetter RD, Goodman CS (2000) Short-range and long-range guidance by Slit and its Robo receptors: a combinatorial code of Robo receptors controls lateral position. Cell 103: 1019–1032
Steenman MJ, Rainier S, Dobry CJ, Grundy P, Horon IL, Feinberg AP (1994) Loss of imprinting of IGF2 is linked to reduced expression and abnormal methylation of H19 in Wilms' tumour. Nat Genet 7: 433–439, (Published erratum appears in Nat Genet 1994; 8: 203)
Stein E, Tessier-Lavigne M (2001) Hierarchical organization of guidance receptors: silencing of netrin attraction by slit through a Robo/DCC receptor complex. Science 291: 1928–1938
Storkel S, van den Berg E (1995) Morphological classification of renal cancer. World J Urol 13: 153–158
Taniguchi T, Sullivan MJ, Ogawa O, Reeve AE (1995) Epigenetic changes encompassing the IGF2/H19 locus associated with relaxation of IGF2 imprinting and silencing of H19 in Wilms tumor. Proc Natl Acad Sci USA 92: 2159–2163
Teitz T, Wei T, Valentine MB, Vanin EF, Grenet J, Valentine VA, Behm FG, Look AT, Lahti JM, Kidd VJ (2000) Caspase 8 is deleted or silenced preferentially in childhood neuroblastomas with amplification of MYCN. Nat Med 6: 529–535
Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP (1999) CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci USA 96: 8681–8686
Tycko B (2000) Epigenetic gene silencing in cancer. J Clin Invest 105: 401–407
Wagner KJ, Cooper WN, Richard G, Grundy RG, Caldwell G, Jones C, Wadey RB, Morton D, Schofield PN, Reik W, Latif F, Maher ER (2002) Frequent RASSF1A tumour suppressor gene promoter methylation in Wilms' tumour and colorectal cancer. Oncogene 21: 7277–7282
Wong K, Ren XR, Huang YZ, Xie Y, Liu G, Saito H, Tang H, Wen L, Brady-Kalnay SM, Mei L, Wu JY, Xiong WC, Rao Y (2001) Signal transduction in neuronal migration: roles of GTPase activating proteins and the small GTPase Cdc42 in the Slit-Robo pathway. Cell 107: 209–221
Xian J, Clark KJ, Fordham R, Pannell R, Rabbitts TH, Rabbitts PH (2001) Inadequate lung development and bronchial hyperplasia in mice with a targeted deletion in the Dutt1/Robo1 gene. Proc Natl Acad Sci USA 98: 15062–15066
Yustein AS, Harper JC, Petroni GR, Cummings OW, Moskaluk CA, Powell SM (1999) Allelotype of gastric adenocarcinoma. Cancer Res 59: 1437–1441
Zöchbauer-Müller S, Fong KM, Virmani AK, Geradts J, Gazdar AF, Minna JD (2001) Aberrant promoter methylation of multiple genes in non-small cell lung cancers. Am J Hum Genet 60: 765–771
Acknowledgements
We thank Cancer Research UK and the Association for International Cancer Research for financial support. TM was supported by the Swedish Cancer Society, the Children's Cancer Foundation, the IngaBritt and Arne Lundberg Foundation, the Assar Gabrielsson Foundation, and the King Gustav V Jubilee Clinic Cancer Research foundation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Astuti, D., da Silva, N., Dallol, A. et al. SLIT2 promoter methylation analysis in neuroblastoma, Wilms' tumour and renal cell carcinoma. Br J Cancer 90, 515–521 (2004). https://doi.org/10.1038/sj.bjc.6601447
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6601447
Keywords
This article is cited by
-
Epigenetics in renal cell cancer: mechanisms and clinical applications
Nature Reviews Urology (2018)
-
Maximal Expression of the Evolutionarily Conserved Slit2 Gene Promoter Requires Sp1
Cellular and Molecular Neurobiology (2016)
-
Reduced expression of Slit2 in renal cell carcinoma
Medical Oncology (2014)
-
SLIT2 inhibits cell migration in colorectal cancer through the AKT–GSK3β signaling pathway
International Journal of Colorectal Disease (2013)
-
Slit-Robo signaling induces malignant transformation through Hakai-mediated E-cadherin degradation during colorectal epithelial cell carcinogenesis
Cell Research (2011)
