Abstract
Increased activation of epidermal growth factor receptor (EGFR) family members such as HER2/Erbb2 can result in more aggressive disease, resistance to chemotherapy and reduced survival of head and neck squamous cell carcinoma (HNSCC) patients. In order to identify mechanisms through which these receptor tyrosine kinases accelerate tumor progression, the regulation of metalloprotease expression by EGFR family members was investigated in 11 squamous cell carcinoma (SCC) cell lines. HER2 expression was significantly correlated with ADAM12 (A Disintegrin And Metalloprotease 12) expression in these cell lines and was co-expressed in human head and neck cancers. Inhibition of HER2 or EGFR decreased ADAM12 transcripts whereas HER2 transfection upregulated ADAM12 expression. To understand the molecular mechanisms underlying HER2 regulation of ADAM12, we investigated the signaling pathways directing ADAM12 production in SCC cells. Inhibition of phosphatidyl inositol-3-kinase or mammalian target of rapamycin decreased ADAM12 transcripts in HER2-expressing SCC cells, whereas transfection with AKT increased ADAM12 mRNA. Experiments utilizing ADAM12 transfection or siRNA targeting of ADAM12 revealed that the protease increased both the migration and invasiveness of oral SCC cells. Surprisingly, ADAM12 also increased HER2 message, protein levels and activity through an Ets1-dependent mechanism. Collectively, these results reveal a novel positive activation loop between ADAM12 and HER2 that may contribute to HNSCC progression.
Similar content being viewed by others


Introduction
Head and neck cancer is the sixth most common cancer worldwide with more than 35 000 new cases annually in the United States alone (Pisani et al., 2002; Jemal et al., 2009). About 90% of these cancers are squamous cell carcinomas (SCCs), and they usually present as locally advanced stage III or IV disease and often metastasize even when identified early (Ragin et al., 2007). Despite extensive research and treatment advances, there has been little improvement in patient 5-year survival rates, which are currently 20–40% for those with locoregionally advanced disease (Chin et al., 2006). One promising treatment option is epidermal growth factor receptor (EGFR) targeted therapeutics (Chin et al., 2006), although little investigation of EGFR's mechanisms or biological functions in oral cancer has been undertaken.
Aberrant expression of EGFR and its dimerization partner HER2 are indicators of poor prognosis in head and neck squamous cell carcinoma (HNSCC). EGFR, overexpressed in 80–90% of HNSCC, is an early event in HNSCC that is associated with more aggressive disease, resistance to chemotherapy and poorer survival (Ibrahim et al., 1997; Forastiere et al., 2001; Ang et al., 2002; Hanawa et al., 2006; Kong et al., 2006; Ettl et al., 2008). The frequency of HER2 overexpression varies between 6% and over 80% depending on tumor type and is associated with shorter disease-free and overall survival (Sato-Kuwabara et al., 2009; Brunner et al., 2010). For these reasons, EGFR and HER2 have been considered appealing targets for cancer therapy. Agents targeting EGFR have been used in a number of clinical trials and are now approved for HNSCC treatment, revealing increased response rates and increased overall survival when combined with standard cytotoxic therapy (reviewed in Moon et al., 2010). Intriguingly, the activation status of HER2 but not EGFR predicts resistance to the EGFR inhibitor gefitinib in HNSCC (Erjala et al., 2006), suggesting that interactions between family members are important for unknown reasons.
EGFR family members can contribute to SCC invasion and progression by upregulating matrix metalloproteinases (MMPs) that digest extracellular matrix, process growth factors and activate cell adhesion molecules (reviewed in Hudson et al., 2009). Tumor cells frequently overexpress MMPs, allowing for degradation of the basement membrane and invasion of the surrounding tissue. In recent years, another family of proteases, the ADAMs (A Disintegrin And Metalloproteinases), have been described and subsequently found to be increased in various human cancers (Kodama et al., 2004; Carl-McGrath et al., 2005; Kveiborg et al., 2005; Lendeckel et al., 2005; Rocks et al., 2006). Among the ADAM family members increased in cancer, ADAM12/Meltrin α is expressed at low levels in most normal adult tissues, and is overexpressed in a large proportion of some human carcinomas, including HNSCC (Carl-McGrath et al., 2005; Kornberg et al., 2005; Roepman et al., 2005; Markowski et al., 2009; Mino et al., 2009). In oral cancers, ADAM12 (A Disintegrin And Metalloprotease 12) upregulation correlates with HNSCC development and progression to metastasis (Kornberg et al., 2005; Roepman et al., 2005; Markowski et al., 2009) although no investigation of its mechanisms of action in head and neck cancer have been previously reported.
ADAM12 is a multifunctional protein with a metalloprotease domain, disintegrin-like region, cysteine-rich domain, transmembrane domain, a prodomain that remains associated with the mature form of the protein and a cytoplasmic tail that can signal through phosphotidyl inositol-3-kinase (PI3K) and other pathways (reviewed in Jacobsen and Wewer, 2009). In humans there are two ADAM12 proteins produced from alternative splicing: ADAM12L, the long form that includes all the domains mentioned, and ADAM12S, the secreted form that lacks the cytoplasmic tail and transmembrane domain but has a unique 33 amino acid tail of unknown function (Jacobsen and Wewer, 2009).
Multiple functions for ADAM12 have been proposed. The protease domain of ADAM12 can cleave extracellular matrix substrates including collagen IV, fibronectin and gelatin (Roy et al., 2004). ADAM12 also causes pathological auto-stimulation of tumor cell growth through proteolytic shedding of EGFR ligands including heparin-binding EGF (HB-EGF) (reviewed in Kveiborg et al., 2008). Other ADAM12 domains appear to have a number of different functions as well. For example, the disintegrin domain of ADAM12L associates with β1 integrins at the plasma membrane to facilitate cell adhesion (Kawaguchi et al., 2003; Thodeti et al., 2005). Whether ADAM12 influences HNSCC progression through these or other mechanisms, and the biological significance of its interactions with receptor tyrosine kinases are currently unknown.
The present study was undertaken to evaluate whether EGFR family members modulate metalloproteinase expression in HNSCC. A correlation between HER2 and ADAM12 expression was discovered in HNSCC cell lines and in human head and neck cancers. Further experiments revealed a positive feedback loop between HER2 and ADAM12 that increases HNSCC migration and invasion. This research reveals a novel mechanism through which HER2 and ADAM12 may contribute to the progression of HNSCC and other cancers.
Results
Increased HER2 expression was associated with ADAM12 expression in human HNSCC cell lines
Eleven human HNSCC cell lines and the control keratinocyte cell line, HaCaT, were used in an initial screen to identify potential associations between metalloproteinases and HER family receptors for further study. Immunoblotting of these lines revealed that most HNSCC cell lines had elevated HER2 compared with the control cells, whereas EGFR and HER3 were not increased in any of the SCC cells (Figures 1a and b). Correlations between HER receptor levels and protease expression were assessed following the measurement of transcripts for seven different MMPs and five different ADAMs (complete list of proteases provided in the Materials and methods section) using real-time RT–PCR. HER2 levels were significantly correlated with ADAM12 transcripts (Figure 1c) and were inversely correlated with MMP14 (R=−0.649, P=0.023, not shown). No significant correlations between EGFR or HER3 and any of proteases were identified. In order to further investigate the relationship between HER2 and ADAM12, the expression of both the prototypic transmembrane form, ADAM12L, and the smaller secreted form, ADAM12S, was determined using primers specific for these splice variants. Expression of ADAM12S transcripts was significantly correlated with HER2 in the HNSCC cells while the association between HER2 and ADAM12L did not reach significance (Figures 1d and e). Immunoblotting for ADAM12 revealed ADAM12 protein in all the cell lines (Figure 1a), with a band at approximately 97 kDa as expected for this antibody (ProteinTech Group, Chicago, IL, USA). Immunohistochemical analysis of human head and neck cancers revealed that all the SCC had detectable HER2, one-third had detectable ADAM12, and all tumors that expressed ADAM12 were also immuno-positive for both EGFR and HER2 (Figure 1f). These data suggested that investigation of a relationship between ADAM12 and HER2 warranted further study.

HER2 levels correlated with ADAM12 expression in head and neck cancer cell lines. (a) Immunoblotting for EGFR family members and ADAM12 in HNSCC cell lines and HaCaT control cells. The blots have been cropped and the lanes reordered so their display is consistent with the ordering of the cell lines in b. (b) Quantification of receptor levels from immunoblots of HNSCC and HaCaT control cell lines. (c–e) HER2 protein levels obtained by quantification of immunoblotting and quantification of ADAM12 (primers that recognize both ADAM12L and ADAM12S), ADAM12L and ADAM12S transcripts following real-time RT–PCR. Values shown (b–e) are the mean for at least two replicates for immunoblotting or are the mean of at least four replicates for real-time RT–PCR. P-values shown are for a test of significance of Pearson's correlation coefficient (R) (c–e). (f) Immunohistochemistry for ADAM12, HER2 and EGFR in a human HNSCC or a negative control lacking primary antibody (far right). Arrows indicate membrane localization of receptors. Scale bar indicates 10 μm in the lower panels. Upper panels are expanded views of the lower panels.
HER2 and ADAM12 expression were increased in oral SCC cells derived from a recurrence compared with cells from the primary tumor from the same site
The expression of ADAM12 and HER2 was evaluated in a primary tumor and from a recurrence at the same site. Cell line UM-SCC 74A is derived from a tongue tumor and UM-SCC 74B is from a tumor recurrence 1 year after the initial biopsy (Worsham et al., 1999). HER2 protein was detected in the recurrent tumor cell line UM-SCC 74B but not in the primary tumor line UM-SCC 74A (Supplementary Figure 1). The message for HER2 was detected in both of these cell lines, although it was significantly increased in the UM-SCC 74B cells (Figure 2a). EGFR protein was also elevated in UM-SCC 74B cells compared with UM-SCC 74A cells, whereas HER3 was not detectable in either line (Figures 1a and b, and Supplementary Figure 1). ADAM12L and ADAM12S transcripts were elevated in the cells from the recurrence when compared with the primary tumor (Figure 2b). Immunofluorescence and immunoblotting confirmed increased ADAM12 protein in the cell line from the recurrent tumor compared with the original tumor cell line (Figures 1a and 2c). ADAM12 immunolocalization in the recurrent tumor cell line was particularly intense in perinuclear regions (Figure 2c arrows).

HER2 and ADAM12 were increased in a recurrent tumor cell line compared with cells from the original primary tumor from the same site. (a, b) Real-time RT–PCR for HER2 (a), ADAM12L (b) and ADAM12S (b) in UM-SCC 74A and UM-SCC 74B cells. *Indicates statistical significance when compared with UM-SCC 74A using a Student's t-test, where P⩽0.05. (c) Immunofluorescence for ADAM12 in UM-SCC 74A and UM-SCC 74B cells. Panels on left show ADAM12 and on right show 4′,6-diamidino-2-phenylindole. Arrows indicate perinuclear ADAM12 localization.
HER2 upregulated ADAM12 expression
To test the hypothesis that HER2 signaling upregulates ADAM12, the low HER2-expressing cell line UM-SCC 74A was transfected with a HER2-mammalian expression construct. Immunoblotting and real-time RT–PCR analyses confirmed increased HER2 following transfection of UM-SCC 74A (Figures 3a and b). HER2 transfection significantly increased transcripts for both ADAM12L and ADAM12S in the UM-SCC 74A cells (Figure 3a). AG825, a potent and relatively specific inhibitor of HER2 (Madson et al., 2006), reduced HER2 activity in UM-SCC 74B cells, as detected by HER2 phosphorylation on immunoblot (Figure 3c). Inhibition of HER2 also reduced ADAM12L and ADAM12S transcripts in a dose-dependent manner, such that ADAM12 transcripts in the high-expressing UM-SCC 74B cell line after treatment with the highest dose were similar to the levels in untreated low-expressing cells (Figure 3d). Collectively, these experiments demonstrated that HER2 induces both ADAM12L and ADAM12S expression in oral SCC cells. Because EGFR and HER2 can heterodimerize to transduce signals, we also investigated whether EGFR influences ADAM12. EGFR activity was inhibited in UM-SCC 74B cells using AG1478, as shown in Figure 3e, lanes 5–8. Inhibition of EGFR also reduced ADAM12 transcripts in UM-SCC 74B cells (Figure 3f), similarly to inhibition of HER2.

HER2 and EGFR upregulated ADAM12 in HNSCC cells. (a, b) The low-ADAM12-expressing cell line UM-SCC 74A was transfected with HER2 or with the empty vector pcDNA. Real-time RT–PCR was performed using primers for ADAM12L, ADAM12S or HER2 (a). HER2 protein was assessed using immunoblotting (b). (c, d) The high-ADAM12-expressing cell line UM-SCC 74B and the low-ADAM12-expressing cell line UM-SCC 74A (as a negative control) were treated with the vehicle DMSO or with the HER2 inhibitor AG825 at the indicated concentrations for 24 h. Phosphorylation of HER2 (P-HER2) on immunoblot (c) and ADAM12L and ADAM12S transcripts following real-time RT–PCR (d) are shown. (e, f) UM-SCC 74A and UM-SCC 74B cells were treated with the EGFR inhibitor AG1478 or with the vehicle DMSO alone. Immunoblotting for phospho-EGFR (P-EGFR)(e) and real-time RT–PCR for ADAM12L and ADAM12S (f) are shown. *Indicates a significant difference compared with the empty vector (a) or vehicle-treated UM-SCC 74B (d, f) controls using a Student's t-test, where P⩽0.05.
HER2 upregulated ADAM12 expression through both PI3K and JNK pathways
To elucidate the mechanisms by which HER2 regulated ADAM12 expression, intracellular signaling pathways downstream from HER2 were examined. Phosphatidyl inositol-3-kinase (PI3K) signaling, as indicated by AKT phosphorylation, was elevated in the high ADAM12 UM-SCC 74B cells when compared with low ADAM12 UM-SCC 74A cells (Figure 4a). Inhibition of PI3K with either LY294002 or wortmannin, confirmed by immunoblotting for phosphorylated AKT (Figure 4a), reduced ADAM12L and ADAM12S transcripts in high ADAM12-expressing UM-SCC 74B cells by 45% and 88%, respectively, to levels similar to that of UM-SCC 74A (Figure 4b). Inhibition of the downstream mammalian target of rapamycin with rapamycin also significantly reduced ADAM12 transcripts (Figure 4c). Conversely, transfection of the low HER2- and low ADAM12-expressing line UM-SCC 74A with a mammalian expression construct for AKT increased ADAM12S and ADAM12L transcripts by more than four-fold (Figure 4d).

PI3K/AKT signaling modulated ADAM12 levels. (a–c) HNSCC cells were treated with the PI3K inhibitors LY294002 (LY) or wortmannin (Wort), with the mammalian target of rapamycin inhibitor rapamycin or with the vehicle DMSO. Immunoblotting for phospho-AKT (P-AKT) is shown 24 h after inhibitor treatment (a). Real-time RT–PCR for ADAM12S and ADAM12L transcripts 24 h after inhibitor treatment is shown (b, c). (d) UM-SCC 74A cells were transfected with an AKT vector 24 h prior to analysis of ADAM12L and ADAM12S via real-time RT–PCR. (e, f) UM-SCC 74A cells were transfected with HER2 and then treated with LY294002 (LY) for 24 h before real-time RT–PCR for HER2 (e) and ADAM12 (f). *Indicates a significant difference compared with the vehicle (b, c, f) or empty vector (d, e) controls using a Student's t-test, where P⩽0.05.
To eliminate the possibility that the effects of PI3K pathway modulation on ADAM12 were the result of altered HER2 transcription (Ou et al., 2008), UM-SCC 74A cells transfected with a HER2 expression vector, in which HER2 expression was driven by a cytomegalovirus promoter, were also treated with the PI3K inhibitor LY294002. The inhibitor did not affect HER2 transcripts but still reduced ADAM12 transcripts (Figures 4e and f), consistent with PI3K modulating ADAM12 transcripts independently of any effect on HER2 expression.
Examination of other pathways downstream from HER2 revealed lesser effects on ADAM12 transcripts. Activation of ERK1/2, JNK and p38 kinase was not increased in UM-SCC 74B cells compared with UM-SCC 74A cells (Supplementary Figures 2A–C). MEK, JNK and P38 kinase inhibitors effectively targeted these pathways (Supplementary Figures 2A–C) and reduced ADAM12 transcripts in SCC 74B cells (Supplementary Figure 2D), although to a lesser extent than blockade of PI3K signaling. Among these inhibitors, inhibition of JNK was the most effective at reducing ADAM12 transcripts, with an approximately 29% and 79% reduction in ADAM12L and ADAM12S transcripts, respectively. In contrast, inhibition of PKC with either rottlerin or GF109203X had no effect on ADAM12 mRNA (Supplementary Figure 2D). Thus, HER2 regulates ADAM12 mRNA through multiple mechanisms, consistent with previous publications showing multiple mechanisms of ADAM12 regulation in other cell types (Le et al., 2003, 2005).
ADAM12 upregulated HER2 expression through Ets1
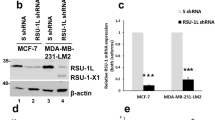
In order to determine whether ADAM12 can modulate the expression and activity of the orphan receptor HER2 in HNSCC cells, we examined the influence of ADAM12 on receptor expression and activity. Transfection of ADAM12L increased HER2 message by more than two-fold in the low-HER2-expressing cell line while transfection with an ADAM12S construct increased HER2 message by 50% (Figures 5a and b). HER2 protein, measured using immunofluorescence, and HER2 activity, measured by immunoblotting for phospho-HER2, were also increased by ADAM12L transfection (Figure 5c). Conversely, siRNA knockdown of ADAM12, which had no effect on other ADAMs (Figure 5d), reduced HER2 message by more than 40% (Figure 5e). Modulation of ADAM12 had no significant effects on EGFR expression (Figures 5b and e).

ADAM12 induced HER2 expression through Ets1 upregulation. (a, b) Low-ADAM12 expressing cell line UM-SCC 74A was transfected with ADAM12L, ADAM12S or an empty vector control and real-time RT–PCR for the indicated genes performed. (c) Cells were transfected with the vector pcDNA or with ADAM12L, as indicated above the panels, and immunofluorescence for HER2 (top) or immunoblotting for phospho-HER2 (P-HER2, bottom) performed. Bar indicates 10 μm. (d, e) High-ADAM12-expressing UM-SCC 74B cells were transfected with ADAM12-targeting or negative control siRNA and real-time RT–PCR performed for the indicated genes. (f, g) Real-time RT–PCR (f) and immunoblotting (g) for Ets1 shown in UM-SCC 74A and UM-SCC 74B cells. (h) Real-time for Ets1 24 h after UMSCC74A cells were transfected with ADAM12L or ADAM12S. (i) Real-time RT–PCR for HER2 performed 24 h after Ets1 or control siRNA transfection in UM-SCC74B cells. *Indicates a significant difference compared with UM-SCC 74A (f), empty vector (a, b, h) or siRNA controls (d, e, i) using a Student's t-test, where P⩽0.05.
To identify mechanisms through which ADAM12 regulates HER2 transcription, a number of transcription factors were examined in the low- and high-ADAM12-expressing cell lines. Ets1 transcripts (Figure 5f) and protein levels (Figure 5g) were elevated in the high-ADAM12-expressing line UM-SCC 74B when compared with the low-ADAM12-expressing UM-SCC 74A. Transfection of cells with ADAM12L or ADAM12S upregulated Ets1 transcripts (Figure 5h), whereas knockdown of Ets1 reduced HER2 transcripts by 60% (Figure 5i). From these data, we conclude that ADAM12 regulation of Ets1 is one mechanism through which this protease upregulated HER2 expression.
ADAM12 increased tumor cell migration and invasion
We hypothesized that upregulation of ADAM12 facilitates tumor progression by increasing tumor cell migration and invasion. Consistent with this hypothesis, migration of high-ADAM12-expressing UM-SCC 74B cells was 67% faster than in the low-ADAM12-expressing UM-SCC 74A cells in a scratch assay (Figure 6a). In addition, knockdown of ADAM12 in UM-SCC 74B significantly decreased cell migration (Figure 6b). Cell invasion through a Matrigel-coated membrane in a Fluoroblok assay was 50% higher in UM-SCC 74B cells when compared with UM-SCC 74A cells (Figure 6c, black bar compared with white bar) and transfection of ADAM12S or ADAM12L increased the invasiveness of UM-SCC 74A cells by 89% and 113%, respectively (Figure 6c, gray bars compared with white bar). These data demonstrate that upregulation of ADAM12 increased both the migration and invasiveness of HNSCC cells.

ADAM12 increased HNSCC cell migration and invasion. (a, b) A scratch assay was performed in UM-SCC 74A and UM-SCC 74B cells (a) or beginning 24 h after transfection of UM-SCC 74B cells with control or ADAM12-targeted siRNA (b). (c) UM-SCC 74A (74A) or UM-SCC 74B (74B) cells were transfected with the empty pcDNA vector or with ADAM12S or ADAM12L and Fluoroblok assays were used to quantify the effect of modulation of ADAM12 on invasion through matrigel. Experiments are representative of at least two performed (a–c). *Indicates significantly different from UM-SCC 74A (a) or from the control siRNA treated (b) using two-way ANOVA or from control transfected UM-SCC 74A using a Student's t-test (c), where P⩽0.05.
Discussion
The present study identified a correlation between ADAM12 and HER2 expression in HNSCC. Further investigation revealed a positive feedback loop between HER2 and ADAM12, which increased the migration and invasiveness of HNSCC cells. In addition, EGFR also positively regulated ADAM12 expression. HER2 induced ADAM12 expression, which in turn upregulated and activated HER2. Thus, there is a positive feedback loop between ADAM12 and HER2 that may have a role in HNSCC progression. Thus, our results suggest that upregulation of ADAM12 expression may be one mechanism through which EGFR or HER2 overexpression results in more aggressive disease.
Understanding the regulation of ADAM12 expression by HER2 in oral SCC tumors may facilitate the development of additional therapeutic interventions for the treatment of tumors. In this regard, we focused on the analysis of HER2-dependent signaling pathways including MAP kinases and PI3K/AKT/mTOR pathways in primary and recurrent SCC tumors. Consistent with other reports demonstrating multiple pathways that regulate ADAM12 expression (Le et al., 2003, 2005), inhibition of each of these kinases decreased ADAM12 transcription to some extent. Inhibition of PI3K, however, was most effective at reducing ADAM12 expression. Because feedback loops exist whereby inhibition of PI3K can downregulate HER2 transcription (Ou et al., 2008), additional experiments were performed to isolate the influence of PI3K inhibition on ADAM12 independent from any effect on HER2. Transfection of HER2 followed by inhibition of PI3K reduced ADAM12 expression to constitutive levels.
ADAM12 has two splice variants, the soluble, short form (ADAM12S) and the long form (ADAM12L) (Jacobsen and Wewer, 2009). HER2 upregulated the expression of both transcripts. Both ADAM12L and ADAM12S have a prodomain, a metalloprotease domain that encodes a viable enzyme, disintegrin and cysteine-rich domains involved in cell adhesion, and EGF-like repeats. They are unique in that the long form also contains a transmembrane region and a cytoplasmic tail, whereas the short form instead has a unique 33 amino-acid sequence (reviewed in Jacobsen and Wewer, 2009). Although both ADAM12 splice variants are frequently cytosolic, ADAM12S is more likely to be secreted whereas ADAM12L can be inserted into the cytoplasmic membrane (reviewed in Jacobsen and Wewer, 2009). Surprisingly, ADAM12L and ADAM12S both induced HER2 expression, although ADAM12L was more effective at increasing HER2 expression.
We found that ADAM12 upregulated HER2 through modulation of the transcription factor Ets1. Ets family regulation of HER2 has previously been reported (Chang et al., 1997; Scott et al., 2000; Xing et al., 2000), although further examination of the mechanisms through which ADAM12 regulation of Ets1 altered receptor expression is required. Ets1 overexpression has been reported in invasive cancers of the oral cavity (Pande et al., 1999) and elsewhere (reviewed in Seth and Watson, 2005). Ets1 is a biological marker of invasive potential and lymph node metastasis in oral SCC and correlates with tumor stage, lymph node involvement, distant organ metastasis and reduced disease-free survival (Pande et al., 2001; Arora et al., 2005). One report suggests that smokeless tobacco extract increases expression of Ets1 and alters the expression of cell cycle regulators in oral leukoplakia (Rohatgi et al., 2005). Ets1 expression correlates with intratumoral microvessel density, VEGF and stromyelysin expression in oral precancerous lesions and SCCs (Arora et al., 2005). Ets1 is an effector of EMT (epithelial to mesenchymal transition) downstream of Snail and TGF-β1 that increases invasiveness of SCC (Shirakihara et al., 2007). Thus, Ets1 may be an important effector of ADAM12-stimulated HNSCC progression through upregulation of HER2 and other mechanisms, a hypothesis warranting further study.
Neither splice variant of ADAM12 had an effect on EGFR transcripts. Both splice variants have proteolytic activity, with distinct substrates identified for each. While ADAM12S cleaves insulin growth factor binding protein-3 and -5, ADAM12L can shed pro-heparin binding epidermal growth factor, resulting in the activation of EGFR (reviewed in Jacobsen and Wewer, 2009). Thus, increased ADAM12L expression may lead to increased EGFR activity by the release of EGFR ligands. The cytoplasmic tail unique to the long form of ADAM12 is involved in both localization of the protease at the cell surface as well as signaling through Src homology 3 (SH3)- and SH2-containing proteins, including Src family members and PI3K p85, suggesting additional and diverse possibilities for ADAM12L signaling (reviewed in Jacobsen and Wewer, 2009). Whether ADAM12L and ADAM12S have distinct roles in HNSCC development and progression, through the activation of EGFR or other mechanisms, remains to be elucidated.
Recently, gene expression analysis has demonstrated elevated ADAM12 in HNSCC tissues correlating with metastasis (Kornberg et al., 2005; Roepman et al., 2005). Our data demonstrated co-expression of ADAM12, HER2 and EGFR in some HNSCCs and that increased ADAM12 in HNSCC cells results in increased HER2 signaling. The frequency of EGFR, HER2 and ADAM12 co-expression in HNSCC in vivo and the prognostic significance of their co-expression, remain to be determined. Increased expression of HER2 has been reported with a poor clinical prognosis in breast cancer patients who have shorter disease-free and overall survival (Slamon et al., 1987), suggesting that HER2 expression may be linked to changes in downstream gene expression leading to increased metastatic ability. HER2 overexpression in some HNSCC is associated with poor clinical prognosis in which patients have shorter disease-free and overall survival (Sato-Kuwabara et al., 2009; Brunner et al., 2010). In addition, EGFR as well as HER2 upregulated the expression of ADAM12. As these receptors can heterodimerize to transduce signals, their individual effects may be due, in part or in whole, to altered EGFR/HER2 dimer-activated signaling. In addition to its effects on HER2 expression in our studies, ADAM12L acts as a sheddase for HB-EGF (reviewed in Jacobsen and Wewer, 2009), and thus may increase EGFR and HER2 activity through an additional mechanism as well. As EGFR is an important therapeutic target for head and neck cancer therapy, ADAM12 may be a potentially important target for HNSCC treatment because of its ability to alter both HER2 and EGFR signaling. Somewhat surprisingly, given the evidence of a strong positive correlation between EGFR and MMP9 in cancers including HNSCC (O-Charoenrat et al., 2000), we found no such correlation in our screen of 11 HNSCC cell lines. Although we can only speculate about the causes for this discrepancy, it may have resulted from the low variation in EGFR protein levels among the HNSCC lines that were used.
In summary, our work demonstrated a positive feedback loop between HER2 and ADAM12 that increases HNSCC cell migration and invasion. Because an inhibitor of ADAM12 is currently in clinical trials (reviewed in Jacobsen and Wewer, 2009), this research suggests that disruption of ADAM12, independently or in conjunction with EGFR/HER2 inhibition, may be a useful target for prevention of HNSCC progression. Further experiments are necessary to elucidate the mechanisms through which ADAM12 acts as well as the biological significance of the events demonstrated here.
Materials and methods
Cell culture
Eleven human HNSCC cell lines (UM-SCC 1, UM-SCC 10A, UM-SCC 11B, UM-SCC 12, UM-SCC 13, UM-SCC 17B, UM-SCC 23, UM-SCC 38, UM-SCC 74A, UM-SCC 74B and UM-SCC 81B) were utilized (Grenman et al., 1991; Squire et al., 2002; Akervall et al., 2004). The cell lines were previously characterized and genotyped as described in (Grenman et al., 1991; Squire et al., 2002; Akervall et al., 2004; Brenner et al., 2010). The cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen, Carlsbad, CA, USA) supplemented with 1% penicillin (10 000 U/ml)-streptomycin (10 000 μg/ml) (Invitrogen) and 10% fetal bovine serum (Gemini Bio-Products, West Sacramento, CA, USA) at 37 °C and 5% CO2. At approximately 80% confluence, this medium was replaced with serum-free DMEM for 24 h before sample collection. Cells were also treated with 10 μM U0126 (Calbiochem, San Diego, CA, USA), 10 μM SB202190 (Calbiochem), 10 μM LY294002 (Calbiochem), 20 μM JNK II inhibitor (SP600125, Calbiochem), 0.5–1.0 μM wortmannin (Calbiochem), AG825 (AG Scientific, San Diego CA, USA), AG1478 (AG Scientific), rapamycin (AG Scientific), 10 μM rottlerin (Calbiochem), 10 μM GF109203X (Calbiochem), or 10 μM SU6656 (Calbiochem) in serum-free DMEM for 24 h. Cells were transfected at 60–80% confluence using Lipofectamine/Plus reagents (Life Technologies, Inc., Carlsbad, CA, USA) according to the manufacturer's protocol with mammalian expression constructs for HER2 (Liu et al., 2001), Akt (plasmid 10841 myrAkt delta4-129, Addgene, Cambridge, MA, USA), Erk1 (plasmid 14440 pcDNA-3-T7-ERK1, Addgene), p38 kinase (plasmid 12658, pMT3P38, Addgene), ADAM12L (Thodeti et al., 2005) and ADAM12S (Thodeti et al., 2005). Transfection efficiency for each experiment was determined by transfection with an enhanced green fluorescent protein (Clontech, Mountain View, CA, USA). Supplementary Figure 3A shows representative green fluorescent protein fluorescence and the mean transfection efficiency of 34%. For siRNA experiments, cells were transfected with siRNA targeting ADAM12 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), Ets1 (Santa Cruz Biotechnology) or with a negative control siRNA (Santa Cruz Biotechnology) using siQuest transfection reagents (Mirus Bio, Madison, WI, USA) according to the manufacturer's instructions. Transfection with Cy3-tagged siRNA was used as a transfection control (Mirus Bio). As shown in Supplementary Figure 3B, the mean transfection efficiency was 91%. Cells were visualized using a Nikon microscope with X10 and X40 objectives, and phase and fluorescence microscopy.
Immunoblotting
Cells were lysed in buffer containing 25 mmol/l Tris-HCl (pH 7.4), 150 mmol/l NaCl, 10% glycerol, 1% Triton-X-100, 1 mmol/l EDTA, complete protease inhibitor cocktail (Roche, Mannheim, Germany), 1 mmol/l Na3VO4, 1.5 μmol/l EGTA, and 10 mmol/l NaF. Protein was assessed by the Bradford assay (Bio-Rad Laboratories, Hercules, CA, USA) using bovine albumin as the standard. Immunoblotting was performed using antibodies recognizing HER2 (Cell Signaling Technology, Inc., Danvers, MA, USA), phospho-HER2 (Cell Signaling Technology, Inc.), EGFR (Santa Cruz Biotech Inc.), phospho-EGFR (Santa Cruz Biotech Inc.), HER2 (Cell Signaling Technology, Inc.), β-actin (Sigma Chemical Co., St Louis, MO, USA), phospho-AKT (Cell Signaling Technology, Inc.), phospho-JNK (Cell Signaling Technology, Inc.), ADAM12 (ProteinTech Group) and p-44/42 MAPK (ERK1/2) (Cell Signaling Technology, Inc.) using standard techniques as we have previously published (El Abaseri et al., 2006; Madson et al., 2009). The evenness of loading and transfer was confirmed by Ponceau S (Sigma Chemical Co.) staining and by actin immunoblotting. Immunoreactive proteins were visualized with horseradish peroxidase-conjugated secondary antibodies (Cell Signaling Technology, Inc.) and enhanced chemiluminescence (Thermo Scientific, Rockford, IL, USA). Densitometry was performed using a Chemidoc XRS Molecular Imager (Bio-Rad Laboratories, Inc.).
Immunostaining
Immunofluorescence and immunohistochemistry were performed using primary antibodies recognizing ADAM12 (Santa Cruz Biotechnology or ProteinTech, Inc.) or HER2 (cell signaling) together with negative controls where the primary antibody was omitted, Alexafluor 488-conjugated secondary antibodies (Invitrogen), and 4′,6- diamidino-2-phenylindole (Invitrogen). Immunohistochemistry of formalin-fixed sections was performed using a horseradish peroxidase-conjugated secondary antibody and diaminobenzidine substrate with hematoxylin counterstaining. Photomicroscopy was performed using a Nikon microscope and cooled camera with Magnifier software (Boyce Scientific, Gray Summit, MO, USA). Scoring of the immunohistochemistry experiments was performed using a semi-quantitative, four-grade (0, 1+ 2+ and 3+) method with the investigator blinded as to the identity of the samples. Cases in which fewer than 10% of the tumor cells were only weakly stained were judged as negative.
RNA extraction and real-time RT–PCR
Total RNA was isolated from cells using an RNeasy Mini Kit (Qiagen Sciences, Valencia, CA, USA) according to the manufacturer's instructions. cDNA was prepared with the first-strand cDNA Synthesis Kit (Invitrogen) using oligo dT primers (Invitrogen). Primers included forward (5′-AGCCGCGAGCACCCAAGT-3′) and reverse (5′-TTGGTGGGCAGGTAGGTGAGTT-3′) primers for HER2, forward (5′-AGCTATGTCTTAGAACCAATGAAAAGTG-3′) and reverse (5′-CCCCGGACGCTTTTCAG-3′) primers recognizing both ADAM12 splice variants, forward (5′-CACCATTGAAAAACTAAGGTGTGTG-3′) and reverse (5′-GAGCCTGACAGGGTTGGAAG-3′) primers specific for the ADAM12L variant, forward (5′-CTGGGCACCTCCCTTCTGT-3′) and reverse (5′-TGCTTCTGCTTGCCGGA-3′) primers specific for the ADAM12S variant and forward (5′-GAAACCTGCCAAGTATGATGAC-3′) and reverse (5′-ACCTGGTCCTCAGTGTAGC-3′) primers for GAPDH. MMP2, MMP3, MMP7, MMP9, MMP12, MMP13, MMP14, ADAM9, ADAM10, ADAM 17 and ADAM19 were also assessed by real-time RT–PCR in the 11 SCC cell lines and HaCaT controls (see Supplementary Table 1 for primer sequences). Real-time RT–PCR was performed in triplicate using SYBR Green Universal MasterMix (PE-Applied Biosystems, Carlsbad, CA, USA) with a first step of 10 min at 95 °C, followed by 40 cycles of amplification (95 °C for 3 s and 60 °C for 30 s) using the 7500 fast Real-Time PCR system (Perkin-Elmer, Foster City, CA, USA). Fold expression relative to controls was determined after normalizing to GAPDH expression.
Migration and invasion assays
Scratch assays to assess cell migration were performed using confluent dishes following plating of 325 000 cells/well in a six-well dish. Some cells were transfected at 80% confluence and the assay performed 24 h later after the cells reached confluence. A wound was created across the confluent dish using a pipette tip, cells were washed with phosphate-buffered saline and then incubated in serum-free DMEM containing 10 μM mitomycin C (Sigma) to prevent cell proliferation. Images were acquired in at least six marked locations at multiple time points using a Nikon phase contrast microscope (Boyce Scientific) with a X10 objective and cell migration assessed using Image J software (NIH Freeware, Bethesda, MD, USA). A stage micrometer was used to convert pixels in the digital images to microns and the area migrated in each field expressed as microns squared. Experiments were performed twice to confirm reproducible results and data from one experiment are shown. The Fluoroblok (Becton-Dickinson, Franklin Lakes, NJ, USA) invasion assays were performed according to the manufacturer's instructions. Cells were transfected at 80% confluence 24 h before staining with 10 μg/ml DilC12 (Becton-Dickinson) in medium with serum for 1 h and then were replated. Fluoroblok assays were performed in triplicate using 50 000 cells plated in each upper well in serum-free DMEM with the bottom chamber containing DMEM with 10% serum to allow measurement of invasion through a matrigel-coated membrane. Fluorescence at 549/565 μm (excitation/emission) was measured using an EnSpire 2300 Multilabel Reader (Perkin Elmer, Waltham, MA, USA). Experiments were performed three times and representative data from one experiment are shown.
Statistical analysis
Analysis of the 11 SCC cell lines in the initial screen for correlations between receptor and protease expression is given as P-values for a test of significance of Pearson's correlation coefficient (R). In other experiments, statistical analyses for comparison of two groups were performed using a Student's t-test, where significance was considered P⩽0.05. For statistical analysis of data collected at multiple time points, two-way ANOVA was used, where P⩽0.05.
References
Akervall J, Kurnit DM, Adams M, Zhu S, Fisher SG, Bradford CR et al. (2004). Overexpression of cyclin D1 correlates with sensitivity to cisplatin in squamous cell carcinoma cell lines of the head and neck. Acta Otolaryngol 124: 851–857.
Ang KK, Berkey BA, Tu X, Zhang HZ, Katz R, Hammond EH et al. (2002). Impact of epidermal growth factor receptor expression on survival and pattern of relapse in patients with advanced head and neck carcinoma. Cancer Res 62: 7350–7356.
Arora S, Kaur J, Sharma C, Mathur M, Bahadur S, Shukla NK et al. (2005). Stromelysin 3, Ets-1, and vascular endothelial growth factor expression in oral precancerous and cancerous lesions: correlation with microvessel density, progression, and prognosis. Clin Cancer Res 11: 2272–2284.
Brenner JC, Graham MP, Kumar B, Saunders LM, Kupfer R, Lyons RH et al. (2010). Genotyping of 73 UM-SCC head and neck squamous cell carcinoma cell lines. Head Neck 32 (4): 417–426.
Brunner K, Fischer CA, Driemel O, Hartmann A, Brockhoff G, Schwarz S . (2010). EGFR (HER) family protein expression and cytogenetics in 219 squamous cell carcinomas of the upper respiratory tract: ERBB2 overexpression independent prediction of poor prognosis. Anal Quant Cytol Histol 32: 78–89.
Carl-McGrath S, Lendeckel U, Ebert M, Roessner A, Rocken C . (2005). The disintegrin-metalloproteinases ADAM9, ADAM12, and ADAM15 are upregulated in gastric cancer. Int J Oncol 26: 17–24.
Chang CH, Scott GK, Kuo WL, Xiong X, Suzdaltseva Y, Park JW et al. (1997). ESX: a structurally unique Ets overexpressed early during human breast tumorigenesis. Oncogene 14: 1617–1622.
Chin D, Boyle GM, Porceddu S, Theile DR, Parsons PG, Coman WB . (2006). Head and neck cancer: past, present and future. Expert Rev Anticancer Ther 6: 1111–1118.
El Abaseri TB, Putta S, Hansen LA . (2006). Ultraviolet irradiation induces keratinocyte proliferation and epidermal hyperplasia through the activation of the epidermal growth factor receptor. Carcinogenesis 27: 225–231.
Erjala K, Sundvall M, Junttila TT, Zhang N, Savisalo M, Mali P et al. (2006). Signaling via ErbB2 and ErbB3 associates with resistance and epidermal growth factor receptor (EGFR) amplification with sensitivity to EGFR inhibitor gefitinib in head and neck squamous cell carcinoma cells. Clin Cancer Res 12: 4103–4111.
Ettl T, Schwarz S, Kleinsasser N, Hartmann A, Reichert TE, Driemel O . (2008). Overexpression of EGFR and absence of C-KIT expression correlate with poor prognosis in salivary gland carcinomas. Histopathology 53: 567–577.
Forastiere A, Koch W, Trotti A, Sidransky D . (2001). Head and neck cancer. N Engl J Med 345: 1890–1900.
Grenman R, Carey TE, McClatchey KD, Wagner JG, Pekkola-Heino K, Schwartz DR et al. (1991). In vitro radiation resistance among cell lines established from patients with squamous cell carcinoma of the head and neck. Cancer 67: 2741–2747.
Hanawa M, Suzuki S, Dobashi Y, Yamane T, Kono K, Enomoto N et al. (2006). EGFR protein overexpression and gene amplification in squamous cell carcinomas of the esophagus. Int J Cancer 118: 1173–1180.
Hudson LG, Moss NM, Stack MS . (2009). EGF-receptor regulation of matrix metalloproteinases in epithelial ovarian carcinoma. Future Oncol 5: 323–338.
Ibrahim SO, Vasstrand EN, Liavaag PG, Johannessen AC, Lillehaug JR . (1997). Expression of c-erbB proto-oncogene family members in squamous cell carcinoma of the head and neck. Anticancer Res 17: 4539–4546.
Jacobsen J, Wewer UM . (2009). Targeting ADAM12 in human disease: head, body or tail? Curr Pharm Des 15: 2300–2310.
Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ . (2009). Cancer statistics, 2009. CA Cancer J Clin 59: 225–249.
Kawaguchi N, Sundberg C, Kveiborg M, Moghadaszadeh B, Asmar M, Dietrich N et al. (2003). ADAM12 induces actin cytoskeleton and extracellular matrix reorganization during early adipocyte differentiation by regulating beta1 integrin function. J Cell Sci 116: 3893–3904.
Kodama T, Ikeda E, Okada A, Ohtsuka T, Shimoda M, Shiomi T et al. (2004). ADAM12 is selectively overexpressed in human glioblastomas and is associated with glioblastoma cell proliferation and shedding of heparin-binding epidermal growth factor. Am J Pathol 165: 1743–1753.
Kong A, Leboucher P, Leek R, Calleja V, Winter S, Harris A et al. (2006). Prognostic value of an activation state marker for epidermal growth factor receptor in tissue microarrays of head and neck cancer. Cancer Res 66: 2834–2843.
Kornberg LJ, Villaret D, Popp M, Lui L, McLaren R, Brown H et al. (2005). Gene expression profiling in squamous cell carcinoma of the oral cavity shows abnormalities in several signaling pathways. Laryngoscope 115: 690–698.
Kveiborg M, Albrechtsen R, Couchman JR, Wewer UM . (2008). Cellular roles of ADAM12 in health and disease. Int J Biochem Cell Biol 40: 1685–1702.
Kveiborg M, Frohlich C, Albrechtsen R, Tischler V, Dietrich N, Holck P et al. (2005). A role for ADAM12 in breast tumor progression and stromal cell apoptosis. Cancer Res 65: 4754–4761.
Le PH, Bonnier D, Wewer UM, Coutand A, Musso O, Baffet G et al. (2003). ADAM12 in human liver cancers: TGF-beta-regulated expression in stellate cells is associated with matrix remodeling. Hepatology 37: 1056–1066.
Le PH, L′Helgoualc′h A, Coutant A, Wewer UM, Baffet G, Clement B et al. (2005). Involvement of the serine/threonine p70S6 kinase in TGF-beta1-induced ADAM12 expression in cultured human hepatic stellate cells. J Hepatol 43: 1038–1044.
Lendeckel U, Kohl J, Arndt M, Carl-McGrath S, Donat H, Rocken C . (2005). Increased expression of ADAM family members in human breast cancer and breast cancer cell lines. J Cancer Res Clin Oncol 131: 41–48.
Liu F, Qi HL, Zhang Y, Zhang XY, Chen HL . (2001). Transfection of the c-erbB2/neu gene upregulates the expression of sialyl Lewis X, alpha1,3-fucosyltransferase VII, and metastatic potential in a human hepatocarcinoma cell line. Eur J Biochem 268: 3501–3512.
Madson JG, Lynch DT, Svoboda J, Ophardt R, Yanagida J, Putta SK et al. (2009). Erbb2 suppresses DNA damage-induced checkpoint activation and UV-induced mouse skin tumorigenesis. Am J Pathol 174: 2357–2366.
Madson JG, Lynch DT, Tinkum KL, Putta SK, Hansen LA . (2006). Erbb2 regulates inflammation and proliferation in the skin after ultraviolet irradiation. Am J Pathol 169: 1402–1414.
Markowski J, Oczko-Wojciechowska M, Gierek T, Jarzab M, Paluch J, Kowalska M et al. (2009). Gene expression profile analysis in laryngeal cancer by high-density oligonucleotide microarrays. J Physiol Pharmacol 60 (Suppl 1): 57–63.
Mino N, Miyahara R, Nakayama E, Takahashi T, Takahashi A, Iwakiri S et al. (2009). A disintegrin and metalloprotease 12 (ADAM12) is a prognostic factor in resected pathological stage I lung adenocarcinoma. J Surg Oncol 100: 267–272.
Moon C, Chae YK, Lee J . (2010). Targeting epidermal growth factor receptor in head and neck cancer: lessons learned from cetuximab. Exp Biol Med (Maywood.) 235: 907–920.
O-Charoenrat P, Rhys-Evans P, Modjtahedi H, Court W, Box G, Eccles S . (2000). Overexpression of epidermal growth factor receptor in human head and neck squamous carcinoma cell lines correlates with matrix metalloproteinase-9 expression and in vitro invasion. Int J Cancer 86: 307–317.
Ou CC, Hsu SC, Hsieh YH, Tsou WL, Chuang TC, Liu JY et al. (2008). Downregulation of HER2 by RIG1 involves the PI3K/Akt pathway in ovarian cancer cells. Carcinogenesis 29: 299–306.
Pande P, Mathur M, Shukla NK, Ralhan R . (1999). Ets-1: a plausible marker of invasive potential and lymph node metastasis in human oral squamous cell carcinomas. J Pathol 189: 40–45.
Pande P, Soni S, Chakravarti N, Mathur M, Shukla NK, Ralhan R . (2001). Prognostic impact of Ets-1 overexpression in betel and tobacco related oral cancer. Cancer Detect Prev 25: 496–501.
Pisani P, Bray F, Parkin DM . (2002). Estimates of the world-wide prevalence of cancer for 25 sites in the adult population. Int J Cancer 97: 72–81.
Ragin CC, Modugno F, Gollin SM . (2007). The epidemiology and risk factors of head and neck cancer: a focus on human papillomavirus. J Dent Res 86: 104–114.
Rocks N, Paulissen G, Quesada CF, Polette M, Gueders M, Munaut C et al. (2006). Expression of a disintegrin and metalloprotease (ADAM and ADAMTS) enzymes in human non-small-cell lung carcinomas (NSCLC). Br J Cancer 94: 724–730.
Roepman P, Wessels LF, Kettelarij N, Kemmeren P, Miles AJ, Lijnzaad P et al. (2005). An expression profile for diagnosis of lymph node metastases from primary head and neck squamous cell carcinomas. Nat Genet 37: 182–186.
Rohatgi N, Kaur J, Srivastava A, Ralhan R . (2005). Smokeless tobacco (khaini) extracts modulate gene expression in epithelial cell culture from an oral hyperplasia. Oral Oncol 41: 806–820.
Roy R, Wewer UM, Zurakowski D, Pories SE, Moses MA . (2004). ADAM 12 cleaves extracellular matrix proteins and correlates with cancer status and stage. J Biol Chem 279: 51323–51330.
Sato-Kuwabara Y, Neves JI, Fregnani JH, Sallum RA, Soares FA . (2009). Evaluation of gene amplification and protein expression of HER-2/neu in esophageal squamous cell carcinoma using Fluorescence in situ Hybridization (FISH) and immunohistochemistry. BMC Cancer 9: 6.
Scott GK, Chang CH, Erny KM, Xu F, Fredericks WJ, Rauscher III FJ et al. (2000). Ets regulation of the erbB2 promoter. Oncogene 19: 6490–6502.
Seth A, Watson DK . (2005). ETS transcription factors and their emerging roles in human cancer. Eur J Cancer 41: 2462–2478.
Shirakihara T, Saitoh M, Miyazono K . (2007). Differential regulation of epithelial and mesenchymal markers by deltaEF1 proteins in epithelial mesenchymal transition induced by TGF-beta. Mol Biol Cell 18: 3533–3544.
Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL . (1987). Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235: 177–182.
Squire JA, Bayani J, Luk C, Unwin L, Tokunaga J, MacMillan C et al. (2002). Molecular cytogenetic analysis of head and neck squamous cell carcinoma: by comparative genomic hybridization, spectral karyotyping, and expression array analysis. Head Neck 24: 874–887.
Thodeti CK, Frohlich C, Nielsen CK, Holck P, Sundberg C, Kveiborg M et al. (2005). Hierarchy of ADAM12 binding to integrins in tumor cells. Exp Cell Res 309: 438–450.
Worsham MJ, Wolman SR, Carey TE, Zarbo RJ, Benninger MS, Van Dyke DL . (1999). Chromosomal aberrations identified in culture of squamous carcinomas are confirmed by fluorescence in situ hybridisation. Mol Pathol 52: 42–46.
Xing X, Wang SC, Xia W, Zou Y, Shao R, Kwong KY et al. (2000). The ets protein PEA3 suppresses HER-2/neu overexpression and inhibits tumorigenesis. Nat Med 6: 189–195.
Acknowledgements
The cell lines used in this study were gifts from Drs Greg Oakley (University of Nebraska, Lincoln) and Thomas Carey (University of Michigan). We also thank Drs U. Wewer and C. Frohlich (University of Copenhagen, Denmark) for the gifts of ADAM12 plasmids. This research was supported by the National Institutes of Health (1RO1ES015585), the State of Nebraska LB595 and The Dobleman Head and Neck Cancer Institute. This investigation was conducted in a facility constructed with support from Research Facilities Improvement Program Grant Numbers 1CO6RR17417-01 and G20RR024001 from the National Center for Research Resources, National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Dr Hansen's work has been funded by the NIH, the Nebraska Cancer and Smoking-Related Diseases Research Program and the Dobleman Institute for Head and Neck Cancer Research. The other authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Oncogene website )
Rights and permissions
About this article
Cite this article
Rao, V., Kandel, A., Lynch, D. et al. A positive feedback loop between HER2 and ADAM12 in human head and neck cancer cells increases migration and invasion. Oncogene 31, 2888–2898 (2012). https://doi.org/10.1038/onc.2011.460
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2011.460
