
Abstract
Tumour-induced expansion of regulatory T (TReg) cells is an obstacle to successful cancer immunotherapy. The potential benefit of TReg-cell depletion through the interleukin-2 receptor is lost by the concurrent elimination of activated effector lymphocytes and possibly by the de novo induction of TReg-cell replenishment. In theory, the functional inactivation of TReg cells will maintain them at high numbers in tumours and avoid their replenishment from the peripheral lymphocyte pool, which has the capacity to further suppress the effector lymphocyte anti-tumour response.
Similar content being viewed by others
Main
Since their discovery in the 1960s as suppressive T cells1, regulatory T (TReg) cells have been extensively studied in a wide range of both physiological and pathological conditions in mouse and man. It is now well established that TReg cells are a distinct lymphocyte lineage endowed with regulatory properties that affect a variety of immune cells in the innate and adaptive, and priming and effector phases of the immune response2,3 (Fig. 1). It is now also widely acknowledged that inhibiting TReg-cell function in patients with cancer is an essential step if new therapies, especially immunotherapies, are to be broadly successful in the clinic. But how should the function of TReg cells be inhibited? Initial investigations have indicated that depleting TReg cells from cancer patients might be a valid approach, but more recent, preliminary data has raised the hypothesis that functionally inactivating TReg cells might be better. This Opinion explores this hypothesis.

Tumour cells produce suppressive factors that directly act on regulatory T (TReg) cells or license myeloid-derived suppressor cells, tumour-associated macrophages and dendritic cells to expand TReg cells. Such expansion occurs through the proliferation of pre-existing TReg cells and the conversion of naive precursors into de novo induced TReg cells. Tumour-associated TReg cells suppress both the innate and the adaptive immune response, including the priming and effector phase of both CD4 (green) and CD8 (brown) T cells. IDO, indoleamine 2,3-dioxygenase; TGFβ, transforming growth factor β.
Extinguishing the T Reg cell 'friendly fire'
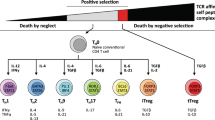
TReg cells are currently classified into two main subsets, according to their origin and suppressive activity4. Natural TReg cells originate in the thymus by high-affinity interaction of the T-cell receptor (TCR) with antigens expressed in the thymic stroma5. They have been classically described to suppress the proliferation of responder T cells in vitro in a contact-dependent, cytokine-independent manner. Natural TReg cells constitutively express CD25, cytotoxic T-lymphocyte antigen 4 (CTLA4), glucocorticoid-induced tumour necrosis factor (TNF) receptor (GITR) and OX40, molecules that characterize activated lymphocytes6,7,8,9,10. However, the high-level expression of the transcription factor FOXP3 (forkhead box p3) is the most distinctive marker for the regulatory lineage, at least in the murine system2. Although incompletely characterized to date, the network of natural TReg-cell suppression mechanisms includes surface molecules such as CTLA4, membrane-bound transforming growth factor β (TGFβ) and the pericellular generation of adenosine3,11,12. Adaptive TReg cells are antigen-specific suppressive cells that are generated in the presence of interleukin 10 (IL10) or TGFβ in case of T regulatory 1 or T helper 3 subsets, respectively13,14. In contrast to natural TReg cells, adaptive TReg cells exert suppression mostly through soluble factors (IL10 and/or TGFβ) and their suppressive function is not strictly associated to high-level FOXP3 expression.
The incompletely understood panoply of TReg-cell-derived immunosuppressive factors regulates the response of nearby cells in a dominant fashion. TReg cells require TCR triggering to become functional, but once activated they suppress T cells blindly, with no regard to the antigen specificity of targeted cells. This implies that the number and function of TReg cells should be tightly controlled to maximize immunity against foreign pathogens and preserve self tolerance. When the number of TReg cells increases, the immune response is unbalanced and favours unresponsiveness. This condition is evident in many murine and human cancers up to the critical point at which tumours are no longer attacked by the immune cells. The available data indicate that TReg-cell expansion can occur through the proliferation of pre-existing TReg cells15,16, and from the conversion of peripheral naive precursors into newly derived TReg cells17,18 (Table 1). Thus, through mechanisms that still need to be examined in detail, tumours turn the host TReg cells from helpful guardians against exacerbated immunity into detrimental suppressors of the anti-tumour immune response.
Several strategies have been proposed to combine TReg-cell neutralization with conventional anti-tumour therapies (Table 2). This might be achieved by targeting TReg-cell development, frequency, migration or activity. The choice of the most suitable approach will depend on the type and stage of tumour, because the mechanisms of immune suppression and the susceptibility to different treatments can vary.
Physiological T Reg -cell homeostasis
TReg cells account for 5–10% of the circulating CD4+ T-cell population. In naive mice, the vast majority of peripheral TReg cells are of thymic origin, as suggested by the observation that the TCR repertoire of peripheral and thymic TReg cells displays a 90% overlap19. However, which antigens are recognized by the TCR repertoire of TReg cells is still poorly understood. Several reports support the idea that natural TReg cells have a diverse TCR repertoire that significantly differs from conventional naive T cells and preferentially recognizes self-antigens19,20,21, in keeping with their exposure to antigens expressed in the thymic stroma5. Self-reactive regulatory thymic emigrants are maintained in the periphery through continuous stimulation by self-peptides expressed at a low level by antigen-presenting cells22. Co-stimulatory molecules, such as B7.1 (also known as CD80) and CD40, are required for peripheral TReg-cell maintenance23,24, and interleukin 2 (IL2) is crucial for TReg-cell homeostasis; IL2-null or IL2 receptor-null mice develop multiorgan fatal autoimmunity that can be reversed by reconstitution with functional TReg cells or by exogenous IL2 (Ref. 25). IL2 also restores TReg-cell number in CD40-deficient mice23. Throughout life, the major role of naturally arising TReg cells is to avoid autoimmunity that is generated from fortuitous activation of autoreactive T-cell clones that have escaped negative selection (deletion of high affinity self-reactive immature T cells) in the thymus. Indeed, both neonatal and adult mice in which TReg cells are acutely ablated succumb to generalized autoimmunity26.
During life, exposure to neo-antigens might enrich the TCR repertoire of TReg cells by conversion, a process in which mature, conventional, non-regulatory CD4+ T cells acquire the regulatory phenotype and gain suppressive functions following peripheral stimulation under certain conditions27. Adoptive transfer of congenic, TCR transgenic or fluorochrome-labelled non-regulatory CD4+ T cells enables their phenotypic changes to be tracked in recipient mice. Such changes include expression of FOXP3 and suppressive functions. These experiments also enable the characterization of the conditions that drive conversion, such as suboptimal antigen stimulation in combination with TGFβ28. TGFβ and retinoic acid-mediated conversion has an important role in establishing intestinal tolerance to exogenous antigens29,30,31. Moreover, recent findings support the notion that peripheral conversion is involved in establishing local immune privilege in transplanted organs32. Therefore, whereas natural TReg cells are centrally induced against the predetermined self-antigen repertoire, TReg cells that developed in the periphery derive from the encounter with any available cognate antigen that is presented in a sub-immunogenic context, regardless of its origin, thus allowing flexible adaptation of host tolerance to external challenges.
Thymus involution occurs late in the mouse lifetime but early, at puberty, in man. This implies that, contrary to mice, the human peripheral pool of TReg cells should decrease with age, unless continuously replenished by peripheral conversion. This has been suggested to occur in elderly people whose effector memory cells turn into memory TReg cells33. This finding predicts that, during the lifespan, the TReg-cell repertoire shifts from the centrally determined self recognition to the peripherally acquired memory of foreign antigens. Whether this hypothesis has evolutionary significance is still unclear.
The tumour perturbs T Reg -cell homeostasis
One of the distinctive features of all living organisms is the ability to react to external stimuli while preserving the stability of the internal environment. This homeostatic property results from continuous adjustments to the dynamic equilibrium, within tolerable limits, between host and foreign challenges. On this basis, the increased level of TReg cells that occurs in the presence of tumours, could be interpreted as the highest level compatible with prolonged tumour–host interaction, allowing the tumour to escape the immune attack but preserving the host from exaggerated and indiscriminate immune suppression. Unfortunately, the benefit of such an equilibrium seems temporary for the host, who eventually succumbs to the tumour burden34.
The mechanisms driving TReg-cell expansion in patients with cancer are not fully understood35. Early studies by Robert North demonstrated that, during tumour progression, the initial development of concomitant immunity is followed, and finally subverted, by the establishment of CD4+ T-cell-mediated immune suppression36. This state of hyporesponsiveness is maintained for up to 28 days after the surgical removal of the primary tumour, as is the persistence of the T-cell population that is responsible for the suppression37. This dual-phase model suggested that effector and suppressor subpopulations might be functionally predominant at different stages of tumour progression. A more recent study in mice with and without tumours has correlated immune status with predominant lymphocyte subsets in lymph nodes. The initial increase of activated effector T cells is followed by a rapid decline that coincides with the increase in the number of TReg cells38. In this model, the subversion of immunity into suppression occurs at the time when TReg cells outnumber effector T cells.
The increase in TReg cells is not unlimited and never exceeds 50% of the CD4+ population17,39,40,41,42,43,44,45. Once established, the upper limit of tumour-induced TReg cells remains constant during the late phase of tumour progression. This has been observed in gastric, colorectal and ovarian carcinomas in which the most prominent increase in TReg cells occurs during the early stages of tumour progression and remains almost unchanged at later stages42,44. Furthermore, TReg-cell alterations in peripheral blood of head and neck cancer patients do not normalize in the absence of evident disease after therapy45, suggesting that post-operative systemic immune suppression might contribute to disease relapse and metastasis.
In addition to analyses that examined overall numbers of TReg cells, several studies have shown that TReg-cell accumulation and compartmentalization can vary among different types of human cancer. In patients with ovarian cancer, TReg cells progressively accumulate within the tumour mass and in the ascitic fluid, and decrease in the draining lymph nodes. TReg cells are recruited to the tumour by CCL22, which is produced by tumour cells and tumour-associated macrophages42. Accumulation of TReg cells at the tumour site might increase suppression locally when TReg-cell expansion has reached the highest possible level. In breast, pancreatic and colorectal tumours, regional lymph nodes remain heavily infiltrated by TReg cells41,43. However, despite variability among tumour types, some common mechanisms involved in TReg-cell amplification can be identified, as described in the following sections.
Proliferation of pre-existing TReg cells. The vast majority of tumour antigens come from self-molecules and so it is conceivable that such an antigenic assortment will preferentially activate natural TReg cells rather than effector T cells. On activation, TReg cells can proliferate while gaining suppressive activity. Tumour-induced TReg-cell proliferation has been reported in several experimental settings15,16,18,46 (Table 1). In the case of influenza haemagglutinin (HA) used as surrogate tumour antigen, TReg cells are the main proliferating cell fraction in the total CD4+ T-cell population expressing HA-specific TCR when transferred into mice with A20-HA expressing mouse lymphoma16. In addition, when mixed with CD4+CD25− (the naive CD4+, non-regulatory T cell, population) from the same TCR-transgenic mice, TReg cells become the prevalent population on transfer into tumour-bearing mice18. These experiments clearly show that the transferred TReg cells proliferate in preference to naive cells in tumour bearing hosts. Which signals drive TReg-cell proliferation in the tumour setting? IL2 is unlikely because few activated effectors or mature dendritic cells can provide it in the context of general immune suppression. Rather, TGFβ is the cytokine that is thought to foster TReg-cell amplification. Both tumour cells directly47 or 'tumour-educated' immune cells can locally produce large amounts of TGFβ (Fig. 1). Some mouse and rat tumours actively induce myeloid immature dendritic cells to secrete TGFβ and this promotes TReg-cellproliferation15. There is also substantial evidence that indicates the involvement of TGFβ in TReg-cell conversion.
De novo TReg-cell induction. The pool of TReg cells in tumour bearers includes newly generated TReg cells in addition to proliferating natural TReg cells (Table 1). Conversion, described above in physiological contexts, is enforced in the presence of tumours such that the size of the pool of TReg cells is increased at the expense of potential effector T lymphocytes. Evidence for tumour driven conversion comes from many mouse and human studies. For example, when HA-specific CD4+CD25− T cells are transferred into tumour-bearing mice, new TReg cells arise with fully suppressive capability16,18. In addition, polyclonal CD4+CD25− T cells that are purified from non TCR-transgenic naive mice convert into TReg cells in the lymphoid organs of tumour-bearing mice17. In this setting, if antigen encounter is required for conversion, the naive T cells should recognize the few tumour-specific non-self molecules or the more abundant tumour-associated self-antigens. The latter possibility suggests that autoreactive clones, that have escaped thymic deletion, are the most likely TReg-cell precursors in tumour-induced peripheral conversion. As in physiological conditions, conversion does not seem to require proliferation, as lymphocytes from mice treated with doses of vinblastine that induce cytostasis convert into TReg cells if transferred into tumour-bearing mice17. This observation implies that targeting TReg-cell proliferation by cyclophosphamide (see below) might be ineffective because conversion would become the dominant mechanism of TReg-cell expansion.
In addition to TGFβ, a suboptimal level of antigen stimulation favours peripheral conversion28. These two conditions are concomitant in the tumour: antigen presentation is impaired, tumour antigens are poorly immunogenic and TGFβ is constantly available. Another molecule that induces TReg-cell conversion is indoleamine 2,3-dioxygenase (IDO), an enzyme that degrades tryptophan leading to tryptophan starvation and the accumulation of kynurenines, which are toxic catabolites of tryptophan (Fig. 1). Tolerogenic dendritic cells, such as the plasmacytoid subset, can show IDO enzymatic activity, thus promoting TReg-cell conversion48. Tumour cells can also directly degrade tryptophan by IDO expression. For instance, the TReg-cell conversion that is induced in vitro and in vivo by A20 lymphoma cells is abrogated in the presence of an IDO inhibitor49. Moreover, IDO expression by human leukaemic cells induces TReg-cell conversion in vitro and correlates with the increase in the number of TReg cells in the peripheral blood of patients with acute myeloid leukaemia49. Therefore, different molecular pathways can contribute to the generation of a suppressive network in which T cells preferentially convert into TReg cells rather than effector T cells.
Less is known about the mechanisms of TReg-cell expansion in cancer patients. Indirect evidence supports the hypothesis of peripheral conversion for TReg-cell expansion. The most expanded TReg-cell subset in patients with multiple myeloma has the naive rather than the memory phenotype that characterizes pre-existing memory TReg cells or effector/memory T cells50. Furthermore, there is evidence that naive TReg cells that have expanded in cancer patients are not recent thymic emigrants, but cells that have already proliferated and colonized the periphery before conversion50.
TReg-cell conversion versus proliferation. In tumour bearers, conversion and proliferation are mutually independent processes. Indeed, T cells do not require proliferation to convert17 and proliferation of natural TReg cells does not limit or increase the rate of conversion18. The relative contribution of conversion to reach and maintain the upper homeostatic level of TReg cells in tumour bearers becomes evident when TReg cells are depleted. In this case converted TReg cells replace most of the pre-existing natural TReg cells, a process with no obvious benefit for the host. In terms of the TCR repertoire, it is conceivable that, beside tumour antigens, any other available antigen, from normal tissues or pathogens, could guide the cognate lymphocytes through conversion into TReg cells rather than into effector T cells. This allows the peripheral generation of TReg cells with a wide TCR repertoire that increases the likelihood of encountering the antigen activating their suppressive activity. Also, conversion redirects T-cell precursors recognizing tumour antigens into TReg cells rather than into T effectors (Fig. 2). In addition, TReg cells that are derived from conversion contain a higher fraction of cells with CD62LlowCD103+ effector or memory phenotype than pre-existing TReg cells17. This subset has recently been shown to rapidly migrate to inflammatory sites and to suppress in vitro proliferation without further TCR triggering, thus potentially representing a ready-to-suppress population51. Because of the above reasons, depletion of tumour-expanded TReg cells could reduce the host immune response further (Fig. 2). If this is shown to be the case in at least some patients with tumours, depletion-based therapies might then need to be replaced by alternative strategies aimed at the functional inactivation of TReg cells.

In naive conditions, natural regulatory T (TReg) cells (round) display an anti-self T-cell receptor (TCR) repertoire (orange). The tumour microenvironment induces the expansion of pre-existing TReg cells and the conversion of naive T cells (oval) into TReg cells, which might have a non-self (green) or self-reactive (pink) TCR. When CD25+ T cells are depleted, conversion accounts for most of the regenerated TReg cells, which now show a wide TCR repertoire.
Depleting T Reg cells: fewer pros than cons
The alpha-subunit of the IL2 receptor CD25, the first surface marker identified on TReg cells, was soon considered a promising target for TReg-cell neutralization in cancer immunotherapy. The availability of the anti-CD25 monoclonal antibody, PC61, enabled the effects of TReg-cell depletion to be tested in several murine cancer models39,52,53,54,55,56 (Table 2). Despite some efficacy39,52,53,54,55,56, intrinsic limitations apply when PC61 is used to treat established tumours. Time-course experiments in two models showed that the efficacy of PC61 is lost as tumours progress40,54. These observations suggest that, in the naive context before tumour challenge, the only CD25+ tumour-specific cells that can be ablated are natural TReg cells. Under these conditions, immunogenic tumours are rejected when transplanted into mice. However, in mice with established tumours, TReg cells and activated effector T cells that express CD25 are depleted by PC61. Therefore, targeting CD25+ cells in situations where the tumour is well established might be ineffective if both regulatory and effector lymphocytes are eliminated40,54. Interestingly, following the injection of anti-CD25 antibody into tumour-free mice, natural FOXP3+CD4+ T cells do not seem to be completely depleted, but instead are maintained at low levels for up to 6 weeks40. In the presence of a growing tumour, however, depletion seems relatively short with TReg cells being replaced in less than 2 weeks17. Depleting TReg cells also raises the possibility of autoimmunity. However, different doses, schedules and experimental settings of PC61 administration indicate that the generation of autoimmunity is variable40,53.
Monoclonal antibodies to human CD25 that are available for clinical use, such as daclizumab, block IL2 and receptor interaction, thus they are used to treat haematological malignancies57. However, none of these antibodies has been tested for their efficacy in the depletion of TReg cells, and daclizumab has been reported to maintain TReg-cell function in recipients of heart transplants58. So far, most studies in humans have used the immunotoxin denileukin diftitox (Ontak), a fusion protein between the IL2 and the diphtheria toxin, to selectively kill lymphocytes expressing the IL2 receptor. The in vivo anti-tumour efficacy is still under preclinical and clinical investigation, and discrepant results have been reported so far. The open issues are the extent to which denileukin diftitox depletes TReg cells and its therapeutic efficacy in treating different types of tumour based on the administration schedule59,60,61,62 (Table 2). A recent observation of autoimmune retinopathy indicates potential localized adverse effects63. Currently, we can speculate that, similar to PC61, denileukin diftitox might eliminate both effector T cells and TReg cells, and this might be followed by peripheral conversion. Moreover, it is possible that triggering of the IL2 receptor might occur through the IL2 portion of the fusion protein64 thus activating rather than depleting TReg cells. Cyclophosphamide is an alkylating agent that profoundly impairs lymphocyte proliferation. Early studies have shown that suppressor lymphocytes from tumour-bearing hosts are susceptible to this drug65. However, single high-dose administration might result in only a transient TReg-cell depletion. This might be corrected by low-dose metronomic administration, which constantly targets TReg cells as they are induced to proliferate66,67,68. In conclusion, the peripheral generation of TReg cells has profound implications for choosing the right therapeutic option.
Alternative ways to target T Reg cells
Inhibition of tumour-specific TReg-cell expansion. The above discussion suggests that TReg-cell elimination might, in some circumstances, be a relatively blind approach that is unable to distinguish between normal and pathological mechanisms of TReg-cell development. Such approaches could potentially leave uncontrolled autoreactive effectors that would damage normal tissues and tumour tissues alike. In theory, the selective targeting of tumour-associated TReg-cell expansion would preserve the physiological TReg-cell context.
This goal could be achieved by inhibiting, for instance, the IDO pathway (Fig. 3). Several preclinical data confirm that the administration of an IDO inhibitor significantly decreases the rate of peripheral conversion and dramatically impairs tumour growth49,69,70.

Depletion of CD25+ cells results in the elimination of both regulatory T (TReg) cells (orange) and activated effector T cells (CD4 green, CD8 brown). Following TReg-cell depletion, conversion rescues a state of immune tolerance. This could be avoided by targeting mechanisms that are involved in conversion, such as indoleamine 2,3-dioxygenase (IDO) and transforming growth factor β (TGFβ). TGFβ blockade could also inhibit TReg-cell proliferation, without affecting pre-existing TReg-cells. Alternatively, TReg-cell functional inhibition inactivates their suppression (white) without inducing a new wave of conversion, thus leading to effective tumour rejection. TLR8, Toll-like receptor 8.
Another possible target is TGFβ, involved in both proliferation and conversion of TReg cells in tumour bearers (Fig. 3). Mice that are genetically engineered to express a dominant negative form of the TGFβ receptor II on lymphocytes show reduced, if not absent, growth of several transplanted tumours15,71, owing to insensitivity of effector T cells to TGFβ suppression and impairment of peripheral TReg-cell generation. A similar effect is seen with TGFβ silencing in tumour cells, which also enables unimpaired triggering of the immune response and impairs tumour engraftment72.
A certain degree of specificity in targeting tumour-associated TReg cells can be obtained by confining the treatment to the tumour. Intratumour injection of a monoclonal antibody that depletes CD4+ lymphocytes induces rejection of late-stage tumours73. Also, the intratumour administration of an antibody triggering GITR eliminates incipient tumours, in the absence of autoimmunity, but this effect seems to be the result of stimulating effector T cells rather than inhibiting TReg-cell function74,75.
An additional possibility is the inhibition of TReg-cell recruitment to the tumour, an approach that has been proposed but not translated into practice yet42.
TReg-cell functional inactivation. Our discussion so far suggests that inhibiting the function of TReg cells so as to prevent TReg-cell conversion would be a viable approach (Table 2). The goal is to reach a situation in which inactive yet physically present TReg cells would still be 'sensed' and 'counted' by the homeostatic system, which although altered in the presence of a tumour still confines TReg-cell expansion to an upper limit (as described above). In theory, TReg-cell inhibition should avoid the new wave of peripherally converted TReg cells (Fig. 3).
TReg cells constitutively express CTLA4 and GITR. CTLA4 blockade or GITR triggering have been shown to reverse immune suppression as a result of TReg-cell function both in vitro and in vivo7,8. However, recent data indicate that a reinterpretation of these initial findings might be required. Although, in experimental inflammatory bowel disease, CTLA4 blockade directly abolishes TReg-cell suppression76, in tumours the same treatment is only partially dependent on TReg-cell inhibition, and is more active if coupled with TReg-cell depletion77. CTLA4 blockade renders effector T cells resistant to TReg-cell suppression, preserves the TReg-cell intrinsic suppressive capacity and expands the pool of TReg cells78. The mechanisms that mediate these effects are unknown. Similarly, the direct inhibition of TReg cells contributes only minimally to the anti-tumour efficacy of anti-GITR antibodies in vivo. Concomitant antibody-mediated GITR triggering has potent adjuvant effects during a tumour vaccination protocol, even if TReg cells have been previously depleted by PC61 (Ref. 75). Unexpectedly, GITR stimulation even expands the population of TReg cells within the tumour-draining lymph nodes75. In summary, neither CTLA4 nor GITR seem the right target to strike at TReg-cell function in cancer immunotherapy.
Instead, the direct stimulation of Toll-like receptor 8 (TLR8) completely reverses TReg-cell-mediated suppression of human cells. Indeed, TReg cells that are pre-treated with the TLR8 ligand poli-G10 no longer inhibit the in vivo CD8+ response to a human tumour cell line that is grown in immunodeficient mice79. This finding indicates that stimulators of the innate immune response, like TLR8 agonists, might concomitantly impair TReg-cell function.
Murine TReg cells constitutively express OX40, a co-stimulatory molecule belonging to the TNF receptor family. Triggering OX40 completely inhibits the suppressive function of TReg cells in vitro9,10 and abolishes their ability to protect mice from experimental graft versus host disease in vivo9. This distinctive property of OX40, coupled with its well-known co-stimulatory function, fostering memory T-cell survival and recall response80, supports the use of OX40-triggering molecules or antibodies in cancer immunotherapy. Accordingly, prevention and successful treatment of murine tumours by an agonistic antibody to OX40 seem to be mainly through the inhibition of TReg-cell-mediated suppression (S.P. and M.P.C., unpublished data).
Finally, the recent discovery that TReg cells possess ectonucleotidase activity that mediates immune suppression through the generation of pericellular immunosuppressive adenosine11,12, indicates that ectoenzyme inhibitors or receptor antagonists, which are able to interrupt the adenosinergic axis, might be a promising therapeutic strategy for tumours. This follows from the observation that genetic or pharmacological inactivation of the A2A adenosine receptor rescues endogenous anti-tumour T-cell responses and induces successful tumour rejection81.
Conclusions and future perspectives
By inducing TReg-cell expansion, the tumour takes advantage of the inhibitory function that these cells exert on all the immune components. We suggest that avoiding the physical elimination of TReg cells would be potentially useful. This approach should prevent the induction of a new wave of peripherally converted TReg cells that are endowed with a wide TCR repertoire that will easily encounter a cognate antigen resulting in their activation. Conversion would also redirect potential effector T cells toward the TReg-cell phenotype. Alternatively, TReg-cell inactivation is a suitable strategy, which would functionally impair TReg-cell suppression without changing the TCR repertoire of the expanded TReg-cell population. Triggering of TLR8 or OX40, and potentially blocking adenosine, might improve the chances of neutralizing TReg-cell immunosuppression in cancer immunotherapy.
References
Gershon, R. K., Carter, R. L. & Kondo, K. On concomitant immunity in tumour-bearing hamsters. Nature 213, 674–676 (1967).
Zheng, Y. & Rudensky, A. Y. Foxp3 in control of the regulatory T cell lineage. Nature Immunol. 8, 457–462 (2007).
Miyara, M. & Sakaguchi, S. Natural regulatory T cells: mechanisms of suppression. Trends Mol. Med. 13, 108–116 (2007).
Bluestone, J. A. & Abbas, A. K. Natural versus adaptive regulatory T cells. Nature Rev. Immunol. 3, 253–257 (2003).
Aschenbrenner, K. et al. Selection of Foxp3+ regulatory T cells specific for self antigen expressed and presented by Aire+ medullary thymic epithelial cells. Nature Immunol. 8, 351–358 (2007).
Takahashi, T. et al. Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: induction of autoimmune disease by breaking their anergic/suppressive state. Int. Immunol. 10, 1969–1980 (1998).
Takahashi, T. et al. Immunologic self-tolerance maintained by CD25+CD4+ regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J. Exp. Med. 192, 303–310 (2000).
Shimizu, J., Yamazaki, S., Takahashi, T., Ishida, Y. & Sakaguchi, S. Stimulation of CD25+CD4+ regulatory T cells through GITR breaks immunological self-tolerance. Nature Immunol. 3, 135–142 (2002).
Valzasina, B. et al. Triggering of OX40 (CD134) on CD4+CD25+ T cells blocks their inhibitory activity: a novel regulatory role for OX40 and its comparison with GITR. Blood 105, 2845–2851 (2005).
Takeda, I. et al. Distinct roles for the OX40–OX40 ligand interaction in regulatory and nonregulatory T cells. J. Immunol. 172, 3580–3589 (2004).
Borsellino, G. et al. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood 110, 1225–1232 (2007).
Deaglio, S. et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 204, 1257–1265 (2007).
Faria, A. M. & Weiner, H. L. Oral tolerance. Immunol. Rev. 206, 232–259 (2005).
Roncarolo, M. G. et al. Interleukin-10-secreting type 1 regulatory T cells in rodents and humans. Immunol. Rev. 212, 28–50 (2006).
Ghiringhelli, F. et al. Tumor cells convert immature myeloid dendritic cells into TGFβ-secreting cells inducing CD4+CD25+ regulatory T cell proliferation. J. Exp. Med. 202, 919–929 (2005).
Zhou, G., Drake, C. G. & Levitsky, H. I. Amplification of tumor-specific regulatory T cells following therapeutic cancer vaccines. Blood 107, 628–636 (2006).
Valzasina, B., Piconese, S., Guiducci, C. & Colombo, M. P. Tumor-induced expansion of regulatory T cells by conversion of CD4+CD25− lymphocytes is thymus and proliferation independent. Cancer Res. 66, 4488–4495 (2006).
Zhou, G. & Levitsky, H. I. Natural regulatory T cells and de novo-induced regulatory T cells contribute independently to tumor-specific tolerance. J. Immunol. 178, 2155–2162 (2007).
Hsieh, C. S., Zheng, Y., Liang, Y., Fontenot, J. D. & Rudensky, A. Y. An intersection between the self-reactive regulatory and nonregulatory T cell receptor repertoires. Nature Immunol. 7, 401–410 (2006).
Hsieh, C. S. et al. Recognition of the peripheral self by naturally arising CD25+ CD4+ T cell receptors. Immunity 21, 267–277 (2004).
Nishikawa, H. et al. Role of SEREX-defined immunogenic wild-type cellular molecules in the development of tumor-specific immunity. Proc. Natl Acad. Sci. USA 98, 14571–14576 (2001).
Lutz, M. B. & Schuler, G. Immature, semi-mature and fully mature dendritic cells: which signals induce tolerance or immunity? Trends Immunol. 23, 445–449 (2002).
Guiducci, C., Valzasina, B., Dislich, H. & Colombo, M. P. CD40/CD40L interaction regulates CD4+CD25+ T reg homeostasis through dendritic cell-produced IL-2. Eur. J. Immunol. 35, 557–567 (2005).
Tang, Q. et al. Cutting edge: CD28 controls peripheral homeostasis of CD4+CD25+ regulatory T cells. J. Immunol. 171, 3348–3352 (2003).
Malek, T. R. & Bayer, A. L. Tolerance, not immunity, crucially depends on IL-2. Nature Rev. Immunol. 4, 665–674 (2004).
Kim, J. M., Rasmussen, J. P. & Rudensky, A. Y. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nature Immunol. 8, 191–197 (2007).
Liang, S. et al. Conversion of CD4+ CD25− cells into CD4+ CD25+ regulatory T cells in vivo requires B7 costimulation, but not the thymus. J. Exp. Med. 201, 127–137 (2005).
Kretschmer, K. et al. Inducing and expanding regulatory T cell populations by foreign antigen. Nature Immunol. 6, 1219–1227 (2005).
Coombes, J. L. et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-β and retinoic acid dependent mechanism. J. Exp. Med. 204, 1757–1764 (2007).
Sun, C. M. et al. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J. Exp. Med. 204, 1775–1785 (2007).
Benson, M. J., Pino-Lagos, K., Rosemblatt, M. & Noelle, R. J. All-trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. J. Exp. Med. 204, 1765–1774 (2007).
Yates, S. F. et al. Induction of regulatory T cells and dominant tolerance by dendritic cells incapable of full activation. J. Immunol. 179, 967–976 (2007).
Vukmanovic-Stejic, M. et al. Human CD4+ CD25hi Foxp3+ regulatory T cells are derived by rapid turnover of memory populations in vivo. J. Clin. Invest. 116, 2423–2433 (2006).
Dunn, G. P., Old, L. J. & Schreiber, R. D. The three Es of cancer immunoediting. Annu. Rev. Immunol. 22, 329–360 (2004).
Zou, W. Regulatory T cells, tumour immunity and immunotherapy. Nature Rev. Immunol. 6, 295–307 (2006).
North, R. J. & Bursuker, I. Generation and decay of the immune response to a progressive fibrosarcoma. I. Ly-1+2− suppressor T cells down-regulate the generation of Ly-1−2+ effector T cells. J. Exp. Med. 159, 1295–1311 (1984).
Bursuker, I. & North, R. J. Generation and decay of the immune response to a progressive fibrosarcoma. II. Failure to demonstrate postexcision immunity after the onset of T cell-mediated suppression of immunity. J. Exp. Med. 159, 1312–1321 (1984).
Hiura, T. et al. Both regulatory T cells and antitumor effector T cells are primed in the same draining lymph nodes during tumor progression. J. Immunol. 175, 5058–5066 (2005).
Ambrosino, E. et al. Immunosurveillance of Erbb2 carcinogenesis in transgenic mice is concealed by a dominant regulatory T-cell self-tolerance. Cancer Res. 66, 7734–7740 (2006).
Betts, G. et al. The impact of regulatory T cells on carcinogen-induced sarcogenesis. Br. J. Cancer 96, 1849–1854 (2007).
Clarke, S. L. et al. CD4+CD25+FOXP3+ regulatory T cells suppress anti-tumor immune responses in patients with colorectal cancer. PLoS ONE 1, e129 (2006).
Curiel, T. J. et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nature Med. 10, 942–949 (2004).
Liyanage, U. K. et al. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J. Immunol. 169, 2756–2761 (2002).
Sasada, T., Kimura, M., Yoshida, Y., Kanai, M. & Takabayashi, A. CD4+CD25+ regulatory T cells in patients with gastrointestinal malignancies: possible involvement of regulatory T cells in disease progression. Cancer 98, 1089–1099 (2003).
Schaefer, C. et al. Characteristics of CD4+CD25+ regulatory T cells in the peripheral circulation of patients with head and neck cancer. Br. J. Cancer 92, 913–920 (2005).
Bui, J. D., Uppaluri, R., Hsieh, C. S. & Schreiber, R. D. Comparative analysis of regulatory and effector T cells in progressively growing versus rejecting tumors of similar origins. Cancer Res. 66, 7301–7309 (2006).
Liu, V. C. et al. Tumor evasion of the immune system by converting CD4+CD25− T cells into CD4+CD25+ T regulatory cells: role of tumor-derived TGF−β. J. Immunol. 178, 2883–2892 (2007).
Fallarino, F. et al. The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor zeta-chain and induce a regulatory phenotype in naive T cells. J. Immunol. 176, 6752–6761 (2006).
Curti, A. et al. Modulation of tryptophan catabolism by human leukemic cells results in the conversion of CD25− into CD25+ T regulatory cells. Blood 109, 2871–2877 (2007).
Beyer, M. et al. In vivo peripheral expansion of naive CD4+CD25high FoxP3+ regulatory T cells in patients with multiple myeloma. Blood 107, 3940–3949 (2006).
Stephens, G. L., Andersson, J. & Shevach, E. M. Distinct subsets of FoxP3+ regulatory T cells participate in the control of immune responses. J. Immunol. 178, 6901–6911 (2007).
Golgher, D., Jones, E., Powrie, F., Elliott, T. & Gallimore, A. Depletion of CD25+ regulatory cells uncovers immune responses to shared murine tumor rejection antigens. Eur. J. Immunol. 32, 3267–3275 (2002).
Jones, E. et al. Depletion of CD25+ regulatory cells results in suppression of melanoma growth and induction of autoreactivity in mice. Cancer Immun. 2, 1 (2002).
Onizuka, S. et al. Tumor rejection by in vivo administration of anti-CD25 (interleukin-2 receptor α) monoclonal antibody. Cancer Res. 59, 3128–3133 (1999).
Shimizu, J., Yamazaki, S. & Sakaguchi, S. Induction of tumor immunity by removing CD25+CD4+ T cells: a common basis between tumor immunity and autoimmunity. J. Immunol. 163, 5211–5218 (1999).
Comes, A. et al. CD25+ regulatory T cell depletion augments immunotherapy of micrometastases by an IL-21-secreting cellular vaccine. J. Immunol. 176, 1750–1758 (2006).
Waldmann, T. A. Daclizumab (anti-Tac, Zenapax) in the treatment of leukemia/lymphoma. Oncogene 26, 3699–3703 (2007).
Vlad, G. et al. Anti-CD25 treatment and FOXP3-positive regulatory T cells in heart transplantation. Transpl. Immunol. 18, 13–21 (2007).
Barnett, B., Kryczek, I., Cheng, P., Zou, W. & Curiel, T. J. Regulatory T cells in ovarian cancer: biology and therapeutic potential. Am. J. Reprod. Immunol. 54, 369–377 (2005).
Dannull, J. et al. Enhancement of vaccine-mediated antitumor immunity in cancer patients after depletion of regulatory T cells. J. Clin. Invest. 115, 3623–3633 (2005).
Attia, P., Maker, A. V., Haworth, L. R., Rogers-Freezer, L. & Rosenberg, S. A. Inability of a fusion protein of IL-2 and diphtheria toxin (Denileukin Diftitox, DAB389IL-2, ONTAK) to eliminate regulatory T lymphocytes in patients with melanoma. J. Immunother. 28, 582–592 (2005).
Mahnke, K. et al. Depletion of CD4+CD25+ human regulatory T cells in vivo: kinetics of Treg depletion and alterations in immune functions in vivo and in vitro. Int. J. Cancer 120, 2723–2733 (2007).
Ruddle, J. B., Harper, C. A., Honemann, D., Seymour, J. F. & Prince, H. M. A denileukin diftitox (Ontak) associated retinopathy? Br. J. Ophthalmol. 90, 1070–1071 (2006).
Attia, P. et al. Selective elimination of human regulatory T lymphocytes in vitro with the recombinant immunotoxin LMB-2. J. Immunother. 29, 208–214 (2006).
North, R. J. Cyclophosphamide-facilitated adoptive immunotherapy of an established tumor depends on elimination of tumor-induced suppressor T cells. J. Exp. Med. 155, 1063–1074 (1982).
Ghiringhelli, F. et al. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol. Immunother. 56, 641–648 (2007).
Hermans, I. F., Chong, T. W., Palmowski, M. J., Harris, A. L. & Cerundolo, V. Synergistic effect of metronomic dosing of cyclophosphamide combined with specific antitumor immunotherapy in a murine melanoma model. Cancer Res. 63, 8408–8413 (2003).
Lutsiak, M. E. et al. Inhibition of CD4+25+ T regulatory cell function implicated in enhanced immune response by low-dose cyclophosphamide. Blood 105, 2862–2868 (2005).
Hou, D. Y. et al. Inhibition of indoleamine 2, 3-dioxygenase in dendritic cells by stereoisomers of 1-methyl-tryptophan correlates with antitumor responses. Cancer Res. 67, 792–801 (2007).
Muller, A. J., DuHadaway, J. B., Donover, P. S., Sutanto-Ward, E. & Prendergast, G. C. Inhibition of indoleamine 2, 3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nature Med. 11, 312–319 (2005).
Gorelik, L. & Flavell, R. A. Immune-mediated eradication of tumors through the blockade of transforming growth factor-beta signaling in T cells. Nature Med. 7, 1118–1122 (2001).
Thomas, D. A. & Massague, J. TGF-β directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 8, 369–380 (2005).
Yu, P. et al. Intratumor depletion of CD4+ cells unmasks tumor immunogenicity leading to the rejection of late-stage tumors. J. Exp. Med. 201, 779–791 (2005).
Ko, K. et al. Treatment of advanced tumors with agonistic anti-GITR mAb and its effects on tumor-infiltrating Foxp3+CD25+CD4+ regulatory T cells. J. Exp. Med. 202, 885–891 (2005).
Ramirez-Montagut, T. et al. Glucocorticoid-induced TNF receptor family related gene activation overcomes tolerance/ignorance to melanoma differentiation antigens and enhances antitumor immunity. J. Immunol. 176, 6434–6442 (2006).
Read, S. et al. Blockade of CTLA-4 on CD4+CD25+ regulatory T cells abrogates their function in vivo. J. Immunol. 177, 4376–4383 (2006).
Sutmuller, R. P. et al. Synergism of cytotoxic T lymphocyte-associated antigen 4 blockade and depletion of CD25+ regulatory T cells in antitumor therapy reveals alternative pathways for suppression of autoreactive cytotoxic T lymphocyte responses. J. Exp. Med. 194, 823–832 (2001).
Quezada, S. A., Peggs, K. S., Curran, M. A. & Allison, J. P. CTLA4 blockade and GM-CSF combination immunotherapy alters the intratumor balance of effector and regulatory T cells. J. Clin. Invest. 116, 1935–1945 (2006).
Peng, G. et al. Toll-like receptor 8-mediated reversal of CD4+ regulatory T cell function. Science 309, 1380–1384 (2005).
Sugamura, K., Ishii, N. & Weinberg, A. D. Therapeutic targeting of the effector T-cell co-stimulatory molecule OX40. Nature Rev. Immunol. 4, 420–431 (2004).
Ohta, A. et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc. Natl Acad. Sci. USA 103, 13132–13137 (2006).
Author information
Authors and Affiliations
Corresponding author
Related links
Rights and permissions
About this article
Cite this article
Colombo, M., Piconese, S. Regulatory T-cell inhibition versus depletion: the right choice in cancer immunotherapy. Nat Rev Cancer 7, 880–887 (2007). https://doi.org/10.1038/nrc2250
Issue Date:
DOI: https://doi.org/10.1038/nrc2250
This article is cited by
-
Responsive biomaterials: optimizing control of cancer immunotherapy
Nature Reviews Materials (2023)
-
Cancer CD39 drives metabolic adaption and mal-differentiation of CD4+ T cells in patients with non-small-cell lung cancer
Cell Death & Disease (2023)
-
Check(point) yourself before you wreck yourself in tumors
Nature Immunology (2023)
-
Immune checkpoint targeting antibodies hold promise for combinatorial cancer therapeutics
Clinical and Experimental Medicine (2023)
-
Indoleamine 2,3-dioxygenase 2 immunohistochemical expression in medullary thyroid carcinoma: implications in prognosis and immunomodulatory effects
BMC Cancer (2022)
