
Abstract
Ixazomib is an oral proteasome inhibitor, approved in USA, Canada, Australia and Europe in combination with lenalidomide and dexamethasone, for the treatment of patients with multiple myeloma who have received at least one prior therapy. We report a population pharmacokinetic model-based analysis for ixazomib that was pivotal in describing the clinical pharmacokinetics of ixazomib, to inform product labelling. Plasma concentration–time data were collected from 755 patients who received oral or intravenous ixazomib in once- or twice-weekly schedules in ten trials, including the global phase III TOURMALINE-MM1 study. Data were analysed using nonlinear mixed-effects modelling (NONMEM software version 7.2, ICON Development Solutions, Hanover, MD, USA). Ixazomib plasma concentrations from intravenous and oral studies were described by a three-compartment model with linear distribution and elimination kinetics, including first-order linear absorption with a lag time describing the oral dose data. Body surface area on the volume of the second peripheral compartment was the only covariate included in the final model. None of the additional covariates tested including body surface area (1.2–2.7 m2), sex, age (23–91 years), race, mild/moderate renal impairment and mild hepatic impairment were found to impact systemic clearance, suggesting that no dose adjustment is required based on these covariates. The geometric mean terminal disposition phase half-life was 9.5 days, steady-state volume of distribution was 543 L and systemic clearance was 1.86 L/h. The absolute bioavailability of an oral dose was estimated to be 58%.
Similar content being viewed by others


Ixazomib pharmacokinetic data from 755 adult patients enrolled across ten studies were adequately described by a three-compartment model with linear distribution and elimination kinetics, including first-order linear absorption with a lag time describing the oral dose pharmacokinetic profile. |
The geometric mean terminal disposition phase half-life was 9.5 days, steady-state volume of distribution was 543 L and systemic clearance was 1.86 L/h. The absolute bioavailability of an oral dose was estimated to be 58%. |
No ixazomib dose adjustment is required based on body surface area (1.2–2.7 m2), sex, age (23–91 years), race, mild or moderate renal impairment, mild hepatic impairment, and smoking status. |
1 Introduction
Ixazomib is the first oral proteasome inhibitor to enter clinical investigation in hematologic and non-hematologic malignancies [1]. It is approved in the USA, Canada, Australia and Europe in combination with lenalidomide and dexamethasone for the treatment of patients with multiple myeloma (MM) who have received at least one prior therapy, based on results from the global phase III, randomised, double-blind, placebo-controlled trial, TOURMALINE-MM1 [2,3,4,5,6]. In the TOURMALINE-MM1 study, ixazomib plus lenalidomide-dexamethasone treatment was associated with a 35% improvement in progression-free survival vs. placebo plus lenalidomide-dexamethasone [5]. Ixazomib is also undergoing phase III investigation in patients with newly diagnosed MM in combination with lenalidomide and dexamethasone (NCT01850524); as single-agent maintenance therapy in MM (NCT02181413 and NCT02312258); and in patients with relapsed/refractory primary systemic amyloidosis in combination with dexamethasone (NCT01659658).
Clinical studies have demonstrated that ixazomib is rapidly absorbed, with a median time to maximum plasma concentration of 1 h post-dose, and exhibits dose-proportional increases in exposure (area under the concentration–time curve [AUC]) [7, 8]. After once-weekly repeat-dose administration, the accumulation ratio for AUC was demonstrated to be approximately 2 [8]. Metabolism by both cytochrome p450 (CYP) enzymes and non-CYP proteins is expected to be the major clearance (CL) mechanism for ixazomib. At clinically relevant ixazomib concentrations, in vitro studies using human cDNA-expressed CYP isozymes showed that no specific CYP isozyme predominantly contributed to ixazomib metabolism [2]. Following administration of a single oral dose of radio-labelled ixazomib to five patients with advanced cancer, 62% of the administered radioactivity was excreted in urine and 22% in feces [2, 9]. In a study examining ixazomib administration after a high-calorie high-fat meal, the rate and extent of ixazomib absorption was decreased, with a 28% reduction in total systemic exposure (AUC) and a 69% reduction in peak plasma concentration, thereby supporting administration at least 1 h before or at least 2 h after food [10]. Additionally, a reduced dose of ixazomib (3 mg) in patients with severe renal impairment and in patients with moderate or severe hepatic impairment is supported by the results of two phase I studies in these special patient populations [11, 12].
Ixazomib has been investigated after both intravenous (IV) and oral administration, and using both once- and twice-weekly dosing schedules, for patients with hematologic and non-hematologic malignancies. In early phase I studies, ixazomib was administered using body surface area (BSA)-based dosing. A population pharmacokinetic (PK) analysis of ixazomib was conducted early in clinical development, based on data from four phase I clinical trials available at the time [13]. This analysis supported the switch from BSA-based dosing to fixed dosing. Since this initial analysis, substantially more PK data have become available from multiple ixazomib clinical trials conducted across global patient populations, including the TOURMALINE-MM1 study [5]. Accordingly, a more robust analysis of all available PK data was performed to identify any patient characteristics that may contribute to variability in the pharmacokinetics of ixazomib. Results from this analysis were pivotal in describing the clinical pharmacokinetics of ixazomib in the US prescribing information and the European Summary of Product Characteristics [2, 6]. Furthermore, these results can be used to guide future drug development and dosing decisions in the ongoing and future clinical investigations of ixazomib across patient populations and indications.
The objectives of this analysis were to develop a population PK model based on available data from relevant phase I, I/II, and III ixazomib studies, including identification and quantification of statistically significant and clinically relevant covariates on population PK parameters, and to estimate individual PK parameters for patients included in the analysis based on the final population PK model.
2 Methods
2.1 Data Collection
Ixazomib plasma concentration–time data were collected from adult patients with advanced hematologic or non-hematologic malignancies (i.e., MM, lymphoma, light-chain amyloidosis or various solid tumours) who had participated in one of ten clinical trials (Table 1; [5, 7, 8, 13,14,15,16,17,18,19]). Full eligibility criteria for study participants are described in each of the original publications. Patients received oral (BSA-based or fixed) or IV (BSA-based) doses of ixazomib. Ixazomib was administered using once-weekly (days 1, 8, and 15 of a 28-day cycle) or twice-weekly (days 1, 4, 8, and 11 of a 21-day cycle) dosing regimens. All procedures performed in studies involving human participants were in accordance with the International Conference on Harmonisation Good Clinical Practice guidelines and appropriate regulatory requirements.
Plasma concentrations of ixazomib were measured pre-dose and at various timepoints post-dose during ixazomib treatment (Supplementary Table 1). Concentrations of ixazomib were measured using a validated liquid chromatography/tandem mass spectrometry assay. A reverse-phase gradient method, running at a flow rate of 0.3 mL/min on a Fortis Phenyl, 2.1 × 50 mm, 5-μm column (Fortis Technologies Ltd, Neston, UK), provided sample stacking and separation for the analytes. Ixazomib and the internal standard (13C9-ixazomib) were ionised in the positive ion spray mode and detected through multiple reaction monitoring of mass transition pairs at 343.1 → 109.0 m/z and 352.1 → 115.0 m/z, respectively. Assay linearity was achieved over a concentration range of 0.5–500 ng/mL for ixazomib, and precision ranged from 1.7 to 6.1% coefficient of variation, with a bias of −4.0 to 2.3% [12].
Patients with at least three PK samples with concentrations above the lower limit of quantification (LLOQ) were included in the analysis. Ixazomib concentrations below the limit of quantification during active treatment were imputed as LLOQ/2 (0.25 ng/mL) or discarded according to the modified M6 method, in which only the first sample of a sequence of samples below the limit of quantification was set to LLOQ/2 and subsequent samples in the sequence below the limit of quantification were discarded [20].
2.2 Population Pharmacokinetic Modeling
Population PK data analysis was performed using NONMEM software (Version 7.2, ICON Development Solutions, Hanover, MD, USA [21]) for non-linear mixed-effects models, running under Perl-speaks-NONMEM (PsN [22] 4.2.0) on a grid of CentOS 7.0 linux servers, and the Intel Fortran compiler, Version 12. Analysis of results was performed using R (Version 3.0.0 or later [23]). The NONMEM models were developed to describe the log-transformed ixazomib concentrations, and estimates were based on the first-order conditional estimation method. The model was developed in a step-wise manner, guided by a pre-specified analysis plan. First, the structural model was optimised by fitting a fixed-effects model to the observed data. Subsequently, the statistical model describing the within- and between-patient variability was developed, resulting in the base model. Finally, statistically significant and clinically relevant covariates were added to the base model to produce the final model. The population PK model was developed using both IV and oral data. Structural model selection proceeded from a one-compartment model to two- and three-compartment models, and was also guided by the results of the first population PK analysis that clearly demonstrated multi-phasic disposition kinetics of ixazomib, which were best described by a three-compartment model [13].
Inter-individual variability was implemented by exponential random-effect models, corresponding to log-normally distributed individual parameters:
where θ i and θ TV,i are the individual and typical values of the parameter, η i is an individual-level random variable sampled from a normal distribution with a mean of zero and variance of ω 2, and represents the ith individual’s deviation from the typical value.
The residual variability was described by an additive error model on the log-transformed concentrations:
where Y ik is the kth observed ixazomib concentration in the ith individual, C ik is the corresponding individual model-predicted concentration, and ε ik is the residual error component sampled from N(0, σ 2). The variance of the residual error was either constant or time dependent [24] with variance defined as a parametric function of time:
where sd(ε) is the standard deviation of the unexplained random variability, parameterized as a function of the standard deviation at the beginning of the treatment period (SD1), the asymptotic standard deviation (SD0) and the rate constant of change in SD (K SD).
Based on typical and individual parameter estimates, the characteristic half-lives associated with a three-compartment model (t 1/2,α, t 1/2,β and t 1/2,γ), as well as the steady-state volume of distribution were derived [25].
2.3 Covariate Analysis
Covariates were tested for statistical significance and clinical relevance, based on the pre-defined list of relevant parameter-covariate relationships (Supplementary Table 2). All covariates were baseline covariates, except concomitant administration of CYP-modulatory drugs, which was included as a time-dependent covariate. Covariates were tested on CL and volume parameters that included a random effect. Model selection was done on the basis of a log-likelihood ratio test by iteratively adding the most significant of the evaluated covariate-parameter relationships until no more statistically significant covariates (at p = 0.01) that also led to a reduction of at least 10% in the corresponding random-effect variance were found. Subsequently, the least significant covariate was eliminated in a backward deletion procedure until no more non-significant covariates (at p = 0.001) were left in the model.
Continuous covariates were included in the population PK model as power functions, while categorical covariates were implemented as factors:
where θ TV,i is the typical parameter for patient i, defined as a function of the population typical value, θ TV,POP and the individual contributions from continuous (X Cont) and binary (X Cat, with values 0 and 1) covariates. θ 1 and θ 2 represent the respective covariate coefficients.
2.4 Model Evaluation
The objective function value (OFV) was used to initially evaluate whether alternative models represented an improvement in overall fit compared with the candidate model. If the alternative model better fit the data, more elaborate diagnostic tools were used to further assess the quality of the model. Alternative models were compared based on the overall model fit (i.e., NONMEM OFV), precision of parameter estimates (% relative standard error), condition number, random-effect and residual error model variances, and based on the following diagnostic tools: goodness-of-fit plots, prediction-corrected visual predictive check (VPC) and bootstrap. In the VPC, quartiles of prediction-corrected observed data were superimposed on the 1000 corresponding simulated datasets to demonstrate the ability of the model to adequately describe the mean trend and variability in the observed data. Model robustness and parameter estimates were assessed by bootstrap based on 1000 resampled datasets (with replacement).
3 Results
3.1 Data Summary
Pharmacokinetic data were available from 755 adult patients, including 632 patients with MM, 80 patients with advanced solid tumours, 28 patients with lymphoma and 15 patients with light-chain amyloidosis (Table 1; [5, 7, 8, 13,14,15,16,17,18,19]); 347 patients were treated in the phase III TOURMALINE-MM1 study, in which patients with relapsed/refractory MM received once-weekly oral ixazomib in combination with lenalidomide and dexamethasone. Patients in studies C16005, C16008, C16013 and a cohort in TB-MC010034 also received ixazomib in combination with lenalidomide and dexamethasone (n = 168); all other studies used single-agent ixazomib (n = 240). Most patients received oral ixazomib (n = 647), but patients in two phase I studies received IV ixazomib (n = 108); 560 patients received weekly dosing and 195 patients received twice-weekly dosing.
A total of 10,199 PK samples were available; 172 (1.7%) were flagged for exclusion from the analysis because of suspected date and/or sampling time recording errors. Of the 354 (3.5%) samples below the limit of quantification, 124 (1.2%) samples were flagged for exclusion, while the remaining 230 samples (2.3%) were simply imputed to LLOQ/2. Given the small number of samples below the limit of quantification, a more complex imputation method (e.g., M3, M4) was not examined as this would have increased the complexity of the model without providing substantial improvements in the model fit. The demographics and baseline characteristics of patients included in the analysis dataset are summarised in Table 2.
Geometric mean and individual dose-normalised PK data obtained from the phase III TOURMALINE-MM1 study are shown in Fig. 1a. Dose-normalised ixazomib concentration–time profiles for phase I and I/II studies with 21-day treatment cycles (twice-weekly ixazomib dosing) and 28-day treatment cycles (weekly ixazomib dosing) are shown in Fig. 1b and c, respectively. In all three plots, the observed ixazomib concentration was normalised by dividing each individual sample concentration by the preceding ixazomib dose. Plots of raw concentrations (not dose normalised) are shown in Supplementary Figure 1.

Geometric mean of dose-normalised ixazomib concentrations observed in: a TOURMALINE-MM1 binned by visit number (open black circles) with individual observations (grey dots) at the actual sampling times. Triangles indicate protocol-specified dose times and numbers indicate the number of PK samples included in each geometric mean calculation. b Cycle 1 in studies with 21-day treatment cycles (TW dosing) binned by protocol-specified sampling time. Triangles indicate protocol-specified dose times. Error bars show 95% CI of the geometric mean. c Cycle 1 in studies with 28-day treatment cycles (W dosing), binned by protocol-specified sampling time. Triangles indicate protocol-specified dose times. Error bars show 95% CI of the geometric mean. CI confidence interval, IV intravenous, PK pharmacokinetic, PO oral, TW twice weekly, W weekly
3.2 Population Pharmacokinetic Model Development
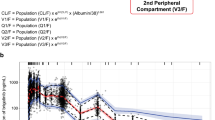
The observed ixazomib plasma concentration–time data after IV or oral administration were adequately described by a three-compartment model with linear distribution and elimination kinetics, including first-order linear absorption with a lag time describing the oral dose PK profile (Fig. 2). The developed model included log-normally distributed patient-level random effects on systemic CL, absolute bioavailability of an oral dose (F) and the volume of the second peripheral compartment (V 4). Individual CL and F were correlated, with coefficient of correlation estimated at 82%. The value of F estimated for patients receiving IV ixazomib was based on the estimated correlation between F and CL. Residual unexplained variability of the log-transformed concentrations was described by an additive error model with time-varying variance described by an exponential decline in the standard deviation (SD) from a starting level (SD1) towards a steady-state plateau (SD0). This residual error model has been previously described by Karlsson et al. [24] and was implemented in the analysis, as a constant additive residual error model showed bias in the conditional weighted residuals and VPCs with respect to time. The SD in the time-varying error model decreases over the first 2–4 h post-dose towards a plateau, reflecting the lower variability in the observed data at later time points.

Structural model describing the pharmacokinetics of ixazomib. ALAG1 absorption lag time, CL clearance, F absolute bioavailability of an oral ixazomib dose, IV intravenous, KA absorption rate constant, Q 3 first inter-compartmental clearance, Q 4 second inter-compartmental clearance, V 2 central volume of distribution; V 3 volume of distribution of first peripheral compartment, V 4 volume of distribution of second peripheral compartment
A covariate model was built based on pre-specified covariate-parameter relationships (Supplementary Table 2). Covariates were only tested on CL and volume parameters that included a random effect. The improvement in OFV, statistical significance, and change in the corresponding inter-individual variances associated with the CL and V 4 parameters following univariate covariate additions are shown in Supplementary Table 3. The addition of univariate covariates corresponded to the first step in the forward addition procedure. Among the parameter-covariate relationships tested, only BSA and body weight met the predefined selection criteria on V 4 by leading to a statistically significant improvement of the model fit, and reducing the random-effect variance by at least 10%. In fact, no other covariates reduced the random-effect variance meaningfully, even when a more conservative criterion of relative reduction of inter-individual variability by ≥5% was used. Because BSA and body weight are strongly correlated body size-related covariates, only one was included in the model. BSA had a small advantage to body weight, both in terms of reducing the OFV and the random-effect variance. Consequently, BSA was included on V 4 as the only covariate in the final model.
The PK parameter values estimated for the final population PK model are presented in Table 3. The final model provided parameter estimates with good precision (relative standard error ≤22%). Furthermore, shrinkage of individual random effects was reasonable (≤37%) with shrinkage of individual CL estimates of 21% in the final model. The geometric mean (95% confidence interval) of the individual estimated terminal disposition phase half-life (t 1/2,γ) was 9.53 (9.32–9.75) days and the steady-state volume of distribution was 543 (534–551) L. These estimates were similar to the population estimates for t 1/2,γ (9.35 days) and steady-state volume of distribution (527 L). The absolute oral bioavailability (F) was estimated to be 58%.
3.3 Pharmacokinetic Model Assessment
To evaluate the appropriateness of the statistical components of the final model, a prediction-corrected VPC was performed [26]. Figure 3 shows a VPC based on data from the first 4 weeks of all studies (Fig. 3a) and from the phase III TOURMALINE-MM1 study (Fig. 3b). Apart from a few time bins, the observed median and 2.5th–97.5th percentiles of data fall within the corresponding 95% confidence interval of the model simulations, indicating that the model adequately describes the central tendency and total variability of the data.

Diagnostic plots: a VPC showing data from the first 4 weeks of all studies based on 1000 simulated datasets. Median and 2.5th–97.5th percentile interval of prediction-corrected observed ixazomib concentrations (black and blue dots, respectively) model simulations (95% CI, grey and blue hatched areas). b VPC showing data from TOURMALINE-MM1 based on 1000 simulated datasets. Median and 2.5th–97.5th percentile interval of prediction-corrected observed ixazomib concentrations (black and blue dots, respectively) model simulations (95% CI, grey and blue hatched areas). c Goodness-of-fit of the final model based on data from all studies (left) and data from TOURMALINE-MM1 (right). Black and grey dots show observed data vs. individual and population predictions, respectively. Red dots indicate observed data below the limit of quantification and the solid line is the line of unity. d Individual random effects on V 4 based on the base model (left) and final model (right) shown vs. body surface area. Solid and dashed lines show a LOESS smooth curve with corresponding 95% CI. BSA body surface area, CI confidence interval, V 4 volume of distribution of second peripheral compartment, VPC visual predictive check
Figure 3c shows the goodness-of-fit plots of the final model based on data from all studies and data from the phase III TOURMALINE-MM1 study. The plots show that both population and individual predictions vs. the observed data scatter randomly around the line of unity, indicating an unbiased fit. As expected, individual predictions are noticeably closer to the line of unity than corresponding population predictions, illustrating the impact of the random-effects model for explaining variability in the observed data. Figure 3d shows the patient-specific random effects (ETA) on V 4 estimated with the base and final models, plotted against individual BSA, and confirms that including BSA as a covariate on V 4 in the final model removes the trend in the random effect on V 4 vs. BSA that was observed with the base model.
Based on the developed final model, the individual predicted exposure following a single oral 4-mg ixazomib dose was calculated using the individual estimated CL and absolute bioavailability as AUC∞ = 4 mg × Fi/CLi. To evaluate trends in individual predicted exposure, AUC∞,i was described using linear regression models including individual covariates as predictors. This corresponds to a graphical covariate screening where individual parameters are plotted vs. covariate values. If no trend is observed, the parameter can be described by the population typical value and random variability. Figure 4 shows the correlation between key continuous covariates and individual predicted exposure and the prediction of the linear regression at the 5th and 95th percentile of continuous covariates relative to the median value. Across the analysis population, the 5th and 95th percentiles of individual predicted exposures were −50 and +98% relative to the median AUC∞. For each continuous covariate, the magnitudes of percent difference in AUC∞ at the 5th or 95th percentiles relative to the AUC∞ at the median of individual covariate values were <20%, and are therefore well below the variability in AUC∞ in the study population. This was consistent with these covariates not being identified as statistically or clinically meaningful covariates on CL. Similarly, as shown in Fig. 5, for categorical covariates, the magnitude of AUC∞ in different categories relative to the most common category were <20% in all cases with the exception of Asian race, where the mean AUC∞ was 35% higher than in White race group. However, there was substantial overlap in exposure between the White and Asian race groups. Taken together, these analyses confirm the absence of clinically meaningful effects of the evaluated covariates on the systemic exposure (AUC∞) of ixazomib in cancer patients.

Correlations between key continuous covariates and individual predicted exposure in patients receiving oral ixazomib. Red and black dots indicate the median and 5th and 95th percentile of individual covariate values, respectively. Numbers (brackets) show the percent change in AUC∞ at the 5th and 95th percentile relative to the value at the median, based on the shown linear regression (and 95% CI). ALB serum albumin, AUC area under the concentration–time curve, BILI total bilirubin, BSA body surface area, CrCl creatinine clearance, HCT hematocrit, WGT weight

Individual predicted exposure stratified by key categorical patient covariates for patients receiving oral ixazomib. Red and black dots indicate the mean exposure in the most prevalent category and in other categories, respectively. Numbers (brackets) in the top of plots show the percent change in AUC∞ (with 95% CI) in other categories relative to the most prevalent category, while numbers at the bottom show patients in each category. AUC area under the concentration–time curve, CI confidence interval, len/dex lenalidomide/dexamethasone
3.4 Model Simulation
The final population PK model was simulated to show the typical concentration–time profile following weekly treatment with oral ixazomib 4 mg in a 28-day treatment regimen (dosing on days 1, 8 and 15). All model parameters were set to the respective typical values with V 4 normalised to the median of individual BSA values reported in the analysis dataset (1.88 m2). Figure 6 shows the simulated concentration–time profile for a typical patient during the first two treatment cycles.

Typical concentration–time profile (and 5th–95th percentile interval of individual predicted profiles) during the first two treatment cycles following oral dosing of ixazomib capsules at 4 mg on days 1, 8 and 15 of 28-day treatment cycles, based on a typical patient (body surface area = 1.88 m2)
4 Discussion
This population PK analysis of the oral proteasome inhibitor ixazomib provides comprehensive model-based integration of data from 755 patients from ten ixazomib clinical trials, including seven phase I studies, two phase I/II studies and one phase III study. The final developed model consisted of a three-compartment structure; oral absorption was described as a first-order linear process with an estimated lag time, while distribution and elimination kinetics were linear. Patient-specific random effects were included on CL, F and V 4. An off-diagonal variance–covariance matrix element representing the correlation between random effects on CL and F was estimated, resulting in considerable improvement in model fit, and indicating a substantial correlation between these two random effects (82%). BSA on V 4 was the only patient covariate included in the final model. None of the other patient covariates tested including BSA, sex, age, race, mild or moderate renal impairment, mild hepatic impairment, smoking status and CYP1A2-modulatory drugs were found to impact CL, suggesting that no dose adjustment is required based on these covariates.
The results presented here are consistent with the previously published population PK analysis of ixazomib, which pooled data from four phase I studies available at the time [13]. In that analysis, ixazomib pharmacokinetics were described by a three-compartment model with first-order absorption and elimination. The estimated absolute bioavailability of ixazomib was 60%, and a small effect of BSA was observed on V 4. The analysis supported a switch from BSA-based to fixed ixazomib dosing without modifications for mild/moderate renal impairment or age. In the present analysis, patients were treated with BSA-based ixazomib doses in studies C16001, C16002, C16003, C16004 and C16005 (phase I part), and fixed ixazomib doses in studies C16005 (phase II part), C16007, C16008, TOURMALINE-MM1, C16013 and TB-MC010034. The present analysis included an additional 529 patients as compared with the previous analysis, and 347 patients were from the phase III TOURMALINE-MM1 study.
Importantly, whereas the first population PK analysis only included studies conducted in the USA/Canada, the present analysis is global in scope, and was inclusive not only of Western, but also Asian patient populations. BSA had a small effect on V 4, but did not impact CL; thereby confirming the results of the previous population PK analysis and further supporting the use of fixed ixazomib dosing. The increased availability of patient level data for the present analysis provided an opportunity to formally evaluate relationships between PK parameters and covariates such as age, baseline bilirubin and creatinine clearance (CrCl) over a wide range of data that included a phase III study and two studies enrolling East Asian patients.
Consequently, the results of the present analysis have additional important implications for the further clinical development of ixazomib and represent pivotal integration of the ixazomib clinical PK database in support of regulatory review and labelling. Age over the range of 23–91 years was not found to have a clinically relevant effect on ixazomib PK parameters, suggesting that no dose adjustments are necessary for elderly patients. This is an important consideration given that the incidence of MM is highest in the elderly [27] and the median age at diagnosis is approximately 69 years [28]. Consistent with the previously published population PK analysis [13], neither CrCl nor total bilirubin had any clinically relevant impact on ixazomib pharmacokinetics, suggesting that no dose adjustments are necessary in patients with mild or moderate renal impairment (CrCl 30–90 mL/min) or patients with mild hepatic impairment (total bilirubin 1–1.5 times the upper limit of normal).
Given that there is a high prevalence of renal impairment in patients with MM [29], a phase I study to assess the pharmacokinetics of ixazomib in patients with severe renal impairment (CrCl < 30 mL/min), including patients with end-stage renal disease requiring dialysis, has been conducted and supports a reduced ixazomib dose of 3 mg in this patient population [11]. Additionally, a phase I study to assess the pharmacokinetics of ixazomib in patients with moderate or severe hepatic impairment (defined as per the National Cancer Institute Organ Dysfunction Working Group criteria) also demonstrated that these patients should receive a reduced dose of 3 mg [12].
Despite the modestly higher AUC in Asian patients (35%), exposures achieved after a 4-mg weekly dose are not expected to exceed those observed at the maximum tolerated dose in Western patients (5.5 mg). Additionally, adverse events following ixazomib treatment are reversible and manageable through dose modification guidelines; hence, no prospective starting dose adjustment is proposed for Asian patients, including Japanese patients. This is also consistent with the PK and safety study conducted in Asian patients in which a 4-mg dose was identified as the recommended phase II/III dose [30].
One of the attributes of this population PK analysis was estimation of absolute bioavailability using PK data collected after IV (n = 108 patients) and oral (n = 647 patients) administration, instead of conducting a dedicated two-way, crossover absolute bioavailability study as required by some regulatory agencies [31]. Use of this model based approach in lieu of a dedicated absolute bioavailability study was accepted by the Australian Government Therapeutic Goods Administration [3].
This population PK analysis included data from patients who received ixazomib as a single agent as well as in combination, allowing the administration in combination with lenalidomide-dexamethasone to be tested as a categorical covariate. There were no alterations in ixazomib pharmacokinetics during combination treatment with lenalidomide-dexamethasone. This is an important finding because ixazomib is approved in combination with lenalidomide-dexamethasone for relapsed/refractory MM. Together with the clinical safety data [5], these results support the continued investigation of ixazomib in combination and as monotherapy in the ongoing phase III clinical trials.
Covariates describing the effect of CYP1A2- and CYP3A4-modulatory concomitant drugs were included in the analysis dataset as time-dependent categorical covariates describing the strongest (most inhibiting or inducing) CYP interaction on each patient-day [32]. Based on covariate analysis, both CYP1A2- and CYP3A4-modulatory drugs, had no statistically significant effect on ixazomib pharmacokinetics. However, it should be noted that a limitation of this aspect of the covariate analysis was that the fraction of records in the analysis dataset that were potentially affected by the CYP-modulatory concomitant drug categories evaluated was very low (1.4 and 3.7% for CYP1A- and CYP3A-modulatory drugs, respectively). Nevertheless, taken together with the established absence of an effect of the strong CYP3A inhibitor clarithromycin on ixazomib pharmacokinetics demonstrated in a controlled phase I clinical study, and the limited contribution of CYP enzymes to the total CL of ixazomib at clinically relevant concentrations [2], these results are consistent with, and support, the conclusion of low risk for clinically relevant drug–drug interactions between ixazomib and co-administered CYP-inhibitory drugs. It should however be noted that the strong pleiotropic PXR-mediated inducer rifampin produced a 74% decrease in systemic exposure (AUC) of ixazomib, likely reflecting an increased contribution of inducible enzymes/disposition mechanisms to the overall CL in the induced state. Accordingly, co-administration of ixazomib with strong CYP3A inducers (such as rifampin, phenytoin, carbamazepine and St. John’s Wort) is not recommended [2].
5 Conclusions
Overall, these results demonstrate how population modeling and simulation can be pivotal in integrating PK knowledge and characterizing sources of variability in systemic exposure, to inform posology decisions and labeling in oncology drug development. Results from this analysis were included in the US Prescribing Information and the European Summary of Product Characteristics for ixazomib.
References
Kupperman E, Lee EC, Cao Y, et al. Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. Cancer Res. 2010;70(5):1970–80.
Millennium Pharmaceuticals Inc. Ninlaro® (ixazomib) capsules, for oral use. United States prescribing information. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2015/208462lbl.pdf. 2015. Accessed 19 Feb 2017.
Australian Register of Therapeutic Goods. Public summary of Ninlaro® ixazomib. Available from: https://www.ebs.tga.gov.au/servlet/xmlmillr6?dbid=ebs/PublicHTML/pdfStore.nsf&docid=260934&agid=(PrintDetailsPublic)&actionid=1. Accessed 19 Feb 2017.
Ninlaro product monograph. Approved by Health Canada. Available from: https://health-products.canada.ca/dpd-bdpp/item-iteme.do?pm-mp=00035983. Accessed 19 Feb 2017.
Moreau P, Masszi T, Grzasko N, et al. Oral ixazomib, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;374(17):1621–34.
European Medicines Agency. Summary of product characteristics: Ninlaro® (ixazomib). Nov 2016.
Richardson PG, Baz R, Wang M, et al. Phase 1 study of twice-weekly ixazomib, an oral proteasome inhibitor, in relapsed/refractory multiple myeloma patients. Blood. 2014;124(7):1038–46.
Kumar SK, Bensinger WI, Zimmerman TM, et al. Phase 1 study of weekly dosing with the investigational oral proteasome inhibitor ixazomib in relapsed/refractory multiple myeloma. Blood. 2014;124(7):1047–55.
Gupta N, Zhang S, Pusalkar S, et al. A phase 1 mass balance study of ixazomib, an oral proteasome inhibitor (PI), using accelerator mass spectrometry (AMS) in patients with advanced solid tumors. American Society for Clinical Pharmacology and Therapeutics Annual Meeting; March 2017: Abstract no. 615.
Gupta N, Hanley MJ, Venkatakrishnan K, et al. The effect of a high-fat meal on the pharmacokinetics of ixazomib, an oral proteasome inhibitor, in patients with advanced solid tumors or lymphoma. J Clin Pharmacol. 2016;56(10):1288–95.
Gupta N, Hanley MJ, Harvey RD, et al. A pharmacokinetics and safety phase 1/1b study of oral ixazomib in patients with multiple myeloma and severe renal impairment or end-stage renal disease requiring haemodialysis. Br J Haematol. 2016;174(5):748–59.
Gupta N, Hanley MJ, Venkatakrishnan K, et al. Pharmacokinetics of ixazomib, an oral proteasome inhibitor, in solid tumour patients with moderate or severe hepatic impairment. Br J Clin Pharmacol. 2016;82(3):728–38.
Gupta N, Zhao Y, Hui AM, et al. Switching from body surface area-based to fixed dosing for the investigational proteasome inhibitor ixazomib: a population pharmacokinetic analysis. Br J Clin Pharmacol. 2015;79(5):789–800.
Suzuki K, Handa H, Chou T, et al. Phase 1 study of ixazomib alone or combined with lenalidomide-dexamethasone in Japanese patients with relapsed/refractory multiple myeloma. Int J Hematol. 2016 Dec 20. Epub ahead of print.
Smith DC, Kalebic T, Infante JR, et al. Phase 1 study of ixazomib, an investigational proteasome inhibitor, in advanced non-hematologic malignancies. Invest New Drugs. 2015;33(3):652–63.
Assouline SE, Chang J, Cheson BD, et al. Phase 1 dose-escalation study of IV ixazomib, an investigational proteasome inhibitor, in patients with relapsed/refractory lymphoma. Blood Cancer J. 2014;4:e251.
Kumar SK, Berdeja JG, Niesvizky R, et al. Safety and tolerability of ixazomib, an oral proteasome inhibitor, in combination with lenalidomide and dexamethasone in patients with previously untreated multiple myeloma: an open-label phase 1/2 study. Lancet Oncol. 2014;15(13):1503–12.
Merlini G, Sanchorawala V, Jeffrey ZA, et al. Long-term outcome of a phase 1 study of the investigational oral proteasome inhibitor (PI) ixazomib at the recommended phase 3 dose (RP3D) in patients (Pts) with relapsed or refractory systemic light-chain (AL) amyloidosis (RRAL). Blood 2014;124(21): Abstract no. 3450.
Hofmeister CC, Rosenbaum CA, Htut M, et al. Twice-weekly oral MLN9708 (ixazomib citrate), an investigational proteasome inhibitor, in combination with lenalidomide (Len) and dexamethasone (Dex) in patients (Pts) with newly diagnosed multiple myeloma (MM): final phase 1 results and phase 2 data. Blood. 2013;122(21):535.
Beal SL. Ways to fit a PK model with some data below the quantification limit. J Pharmacokinet Pharmacodyn. 2001;28(5):481–504.
Beal SL, Sheiner LB. NONMEM users guide: part I–VIII. San Francisco (CA): University of California at San Francisco, NONMEM Project Group C255; 1988.
Lindbom L, Pihlgren P, Jonsson EN. PsN-Toolkit: a collection of computer intensive statistical methods for non-linear mixed effect modeling using NONMEM. Comput Methods Programs Biomed. 2005;79(3):241–57.
R Development Core Team. R: A language and environment for statistical computing. Vienna: R Foundation for Statistical Computing. Available from: http://www.R-project.org/. Accessed 15 Aug 2015.
Karlsson MO, Beal SL, Sheiner LB. Three new residual error models for population PK/PD analyses. J Pharmacokinet Biopharm. 1995;23(6):651–72.
Upton RN. Calculating the hybrid (macro) rate constants of a three-compartment mamillary pharmacokinetic model from known micro-rate constants. J Pharmacol Toxicol Methods. 2004;49(1):65–8.
Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J. 2011;13(2):143–51.
Quach H, Prince HM, Spencer A. Managing multiple myeloma in the elderly: are we making progress? Exp Rev Hematol. 2011;4(3):301–15.
NIH National Cancer Institute. SEER cancer statistics factsheets: myeloma. Available from: http://seer.cancer.gov/statfacts/html/mulmy.html. Accessed 15 Aug 2016.
Dimopoulos MA, Terpos E, Chanan-Khan A, et al. Renal impairment in patients with multiple myeloma: a consensus statement on behalf of the International Myeloma Working Group. J Clin Oncol. 2010;28(33):4976–84.
Gupta N, Goh YT, Min CK, et al. Pharmacokinetics and safety of ixazomib plus lenalidomide-dexamethasone in Asian patients with relapsed/refractory myeloma: a phase 1 study. J Hematol Oncol. 2015;8:103.
Australian Government. Department of Health Therapeutic Goods Administration. Guidance 15: biopharmaceutic studies. 2015; Version 1.1.
Bonate PL, Ahamadi M, Budha N, et al. Methods and strategies for assessing uncontrolled drug-drug interactions in population pharmacokinetic analyses: results from the International Society of Pharmacometrics (ISOP) Working Group. J Pharmacokinet Pharmacodyn. 2016;43(2):123–35.
Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16(1):31–41.
Acknowledgements
The authors thank all patients included in this study and their families, as well as all physicians, nurses, study coordinators and study centre staff participating in the study. The authors also acknowledge Jane Saunders and Helen Wilkinson of FireKite (an Ashfield Company, part of UDG Healthcare plc), who provided medical writing assistance during the development of this manuscript, which was funded by Millennium Pharmaceuticals, Inc., Cambridge, MA, USA, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited and complied with Good Publication Practice 3 ethical guidelines (Battisti et al., Ann Intern Med 2015;163:461–4).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was funded by Millennium Pharmaceuticals, Inc., Cambridge, MA, USA, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited.
Conflict of Interest
NG, MJH, DB, HvdV and KV are employed by Millennium Pharmaceuticals, Inc., Cambridge, MA, USA, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited. PMD is an employee of Quantitative Solutions and contributed as a paid consultant to Millennium Pharmaceuticals, Inc., Cambridge, MA, USA. RDH’s institution received grants for research funding for clinical trial conduct and RDH received an honorarium from Takeda for participating in an advisory board.
Ethics Approval
All procedures performed in studies involving human participants were in accordance with the International Conference on Harmonisation Good Clinical Practice guidelines and appropriate regulatory requirements.
Consent to Participate
All patients provided signed informed consent.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Gupta, N., Diderichsen, P.M., Hanley, M.J. et al. Population Pharmacokinetic Analysis of Ixazomib, an Oral Proteasome Inhibitor, Including Data from the Phase III TOURMALINE-MM1 Study to Inform Labelling. Clin Pharmacokinet 56, 1355–1368 (2017). https://doi.org/10.1007/s40262-017-0526-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-017-0526-4