
Abstract
Background
Helicobacter pylori-cytotoxin-associated protein A (CagA) and gastrin are believed to play an important role in gastric carcinogenesis, but their interaction has been incompletely clear.
Methods
We constructed a eukaryotic expression vector pcDNA3.1/cagA and a luciferase reporter vector pGL/gastrin promoter, and then co-transfected them into gastric cancer AGS and SGC-7901 cells. The two kinds of gastric cancer cells were, respectively, infected with cagA-positive H. pylori NCTC11637, and then the gastrin promoter activity and gastrin mRNA level were detected with a Dual-Luciferase reporter assay system and quantitative reverse transcription polymerase chain reaction (RT-PCR), respectively. Next, after the MEK/ERK and JAK2-signaling pathway inhibitors, U0126 and AG490, were used to treat the two cell lines, or the ERK1 and JAK2 genes were knocked down by siRNAs (small interference RNAs) in the two cell lines, the gastrin promoter activity and gastrin mRNA level were observed again.
Results
The results indicated that CagA could activate the gastrin promoter and up-regulate gastrin mRNA expression in AGS and SGC-7901 cells, but these effects could be inhibited by the inhibitors U0126 and AG490, and the CagA-induced gastrin mRNA expression was down-regulated in the cells whose ERK1 or JAK2 gene was knocked down.
Conclusion
Gastrin promoter may be the transcriptional target of CagA, and CagA activates the gastrin promoter to up-regulate gastrin mRNA expression through the MEK/ERK and JAK1-signaling pathway in gastric cancer cells.
Similar content being viewed by others
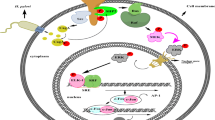


Introduction
Helicobacter pylori-cytotoxin-associated protein A (CagA) and gastrin are believed to play an important role in gastric carcinogenesis [1]. Gastrin, a kind of trophic factor, is produced by gastrointestinal cancer cells and can promote cell proliferation and differentiation [2–4]. Recently, it has been found that gastrin can up-regulate the gene expression of trefoil family factor (TFF)-2 to maintain and repair the gastrointestinal mucosa [3].
Infection with H. pylori is closely related to gastric cancer. CagA, a kind of functional protein and a virulence factor of gastric cancer, is encoded by the cag pathogenicity island (cagPAI) of H. pylori. After the human stomach is infected with H. pylori, CagA is translocated into gastric epithelial cells by the bacterial type IV secretion system, inducing responses of many cells in a tyrosine–phosphorylation-dependent and -independent manner [4].
Recently, there have been some studies about the relationship between serum CagA and gastrin in patients with gastric cancer. Their results show that infection with H. pylori which secretes CagA can significantly raise the risk of gastric cancer, and positive rates of serum CagA and gastrin are all increased in patients with gastric cancer, demonstrating that these entities may play a role in the pathogenesis of gastric cancer [5]. Furthermore, it has been confirmed that H. pylori can induce serum hypergastrinemia and high concentrations of gastrin in patients infected with H. pylori [6]. However, to date, the relationship between H. pylori infection and gastrin has been unclear.
Some evidence has suggested that the activation of signaling plays a critical role in the development of gastric cancer. CagA can also activate multiple signaling pathways including the MEK/ERK and JAK signaling pathways [7]. Therefore, the purpose of this study was to elucidate whether and how H. pylori CagA can inducegastrin expression.
Materials and methods
Cell culture
The human gastric cancer AGS (ATCC CRL-1739™) and SGC-7901 cell lines (from the Cell Bank of Chinese Academy of Sciences, Shanghai, China) were incubated in RPMI-1640 medium (Gibco, Grand Island, NY, USA) supplemented with 10% heat-inactivated fetal bovine serum (Gibco), 100 U/ml of penicillin, and 100 μg/ml of streptomycin at 37°C in a humidified incubator (NSE, Brunswick, NJ, USA) containing 5% CO2.
H. pylori culture
H. pylori NCTC11637 (ATCC 43504, cagA-positive, from the Chinese Center of Helicobacter pylori strain Management and Preservation) was cultured on a Columbia agar plate (Oxoid Ltd, Basingstoke, Hampshire, England) containing 10% sheep blood for 48–72 h at 37°C in a microaerophilic atmosphere. Other H. pylori strains were boiled for 15 min as a negative control that was termed D-Hp.
Construction of the eukaryotic expression vector pcDNA3.1/cagA and luciferase reporter vector pGL/gastrin promoter
DNA of H. pylori NCTC11637 was extracted with proteinase K and phenol. The full-length cagA sequence was synthesized by polymerase chain reaction (PCR), and then cloned into pMD18-T plasmids to construct the recombinant vector pMD18-T/cagA, followed by sequencing. After the pMD18-T/cagA was digested with restriction enzymes PstI and BamHI, cagA was connected to pcDNA3.1/Zeo (−) (Invitrogen, Carlsbad, CA, USA) to construct the eukaryotic expression vector pcDNA3.1Zeo (−)/cagA that was termed pcDNA3.1/cagA. Similarly, the promoter fragment of gastrin was synthesized by PCR with human gastric cancer tissue as DNA template, and then directly cloned into pGL-3 basic vector to construct the reporter vector pGL/gastrin promoter that was termed pGL/GP. The primers used in this study are listed in Table 1.
H. pylori NCTC11637 infection
After gastric cancer AGS and SGC-7901 cells were routinely incubated in 6-well plates containing antibiotic-free medium for 40 h, the inhibitor AG490 (40 μM; Sigma, St. Louis, MO, USA) of the JAK2 signaling pathway and the inhibitor U0126 (10 μM; Sigma) of the MEK/ERK signaling pathway were, respectively, added to the two cell lines to treat the cells for 2 h. Next, these cells were infected with H. pylori NCTC11637 with a multiplicity of infection (MOI) of 100. Six hours later, these cells were harvested for detecting gastrin mRNAs.
Transfection of pcDNA3.1/cagA and pGL/GP
After the AGS and SGC-7901 cells had been incubated in 6-well plates for 12 h, 80% confluent cells occurred. These cells were transfected or co-transfected, according to the manufacturer’s instructions, with 2 μg of DNA of each plasmid and in the presence of 4 μl of Lipofectamine 2000 (Invitrogen) per well. Six hours later, the medium was replaced by antibiotic-free medium containing 10% fetal bovine serum, and then the inhibitors AG490 (40 μM) and U0126 (10 μM) were, respectively, added to the AGS and SGC-7901 cells to treat the cells for 48 h. Finally, the cells were harvested for detecting gastrin. The pcDNA3.1/Zeo (−) and Renilla luciferase reporter plasmid pRL-TK (Clontech, Mountain View, CA, USA) served as negative and internal controls, respectively.
pGL/GP transfection and subsequent H. pylori NCTC11637 infection
After co-transfection with pGL/GP and pRL-TK for 24 h as described above, the cells were infected with H. pylori NCTC11637 twice, every 12 h. Forty eight hours after the last infection, the cells were harvested.
Quantitative reverse transcription (RT)-PCR for gastrin mRNA
Total RNA was extracted from 1 × 106 of cells with TRIzol (Invitrogen) reagent and treated with DNase I (RNase-free) according to the manufacturer’s instructions. Five μg of total RNA was reverse transcribed (RT) with MMLV reverse transcriptase and oligo (dT)15 primers (Promega, San Luis Obispo, CA, USA). Complementary DNA was submitted to real-time quantitative PCR with a TaqMan® Gastrin Gene Expression Assay kit (ABI (Scoresby, Victoria, Australia); Assay ID Hs00174945_m1), and Human ACTB (beta-actin) served as the endogenous control (ABI; Part Number 4333762T). Crossing threshold (C t) values of the gastrin gene were normalized to beta-actin, and then the data were calculated and analyzed by a comparative C t method [8]. Gastrin mRNA expression was expressed as a percentage relative to the control.
Protein extraction and western blotting
The harvested cells were washed with phosphate-buffered saline (PBS), and then added to 100 μl of assay buffer containing protease inhibitor cocktail (Amresco, Solon, OH, USA). Protein concentrations were determined by the Bradford method. Thirty μg of protein was loaded on 8% sodium dodecylsulfate (SDS)-polyacrylamide separating gels and run at 110 V for 2 h at room temperature. Proteins were then transferred onto a polyvinylidene difluoride (PVDF) membrane (Millipore, Billerica, MA, USA). After sealing, the membranes were incubated overnight with rabbit polyclonal anti-CagA antibody (1:800; Santa Cruz Biotechnology, Santa Cruz, CA, USA), mouse monoclonal anti-phosphotyrosine antibody (PY99, 1:300; Santa Cruz Biotechnology), rabbit polyclonal anti-JAK2 antibody (1:500; Santa Cruz Biotechnology), rabbit polyclonal anti-ERK1/2 antibody (1:1000; CangChen, Shanghai, China), and mouse monoclonal anti-glyceraldehyde-3-phosphate-dehydrogenase (GAPDH) antibody (1: 8000; CangChen) in TBS+Tween 20 milk, respectively. The blots were then incubated with the corresponding secondary horseradish peroxidase-conjugated goat anti-rat IgG or goat anti-mouse IgG (1:5000; Santa Cruz Biotechnology) for 2 h at room temperature, respectively. Bands were visualized with the chemiluminescence reagent ECL Plus (Amersham Biosciences, Buckinghamshire, UK) using hyperfilm (Amersham Biosciences).
Luciferase reporter gene assay
Twenty-four hours after co-transfection with the vector pGL/GP and control vector pRL-TK, the AGS and SGC-7901 cells were encoded with firefly and Renilla luciferase, respectively, and then transfected with pcDNA3.1/cagA or infected with H. pylori NCTC11637. Meanwhile, the inhibitors AG490 and U0126 were added to the cells. After 48 h, the cells were harvested and the luciferase activity in cell lysates was detected with the Dual-Luciferase Reporter Assay System (Promega). The vector pRL-TK served as an internal control. The luciferase activity was normalized to Renilla luciferase.
Knockdown of JAK2 and ERK1 genes
The siRNA (small interference RNA) sequences were as follows: JAK2: 5′-CAAGAGGGUUCAA AUGAAATT-3′; ERK1: 5′-GGAUCAGCUCAACCACAUUTT-3′; scraped siRNA: 5′-UUCUCCG AACGUGUCACGUTT-3′ (Shanghai Gene Pharma, Shanghai, China). For the infection experiment, after transfection with JAK2-siRNA (50 nM) and ERK1-siRNA (50 nM), respectively, in the presence of 6 μl Lipofectamine 2000 for 42 h the AGS and SGC-7901 cells were infected with H. pylori NCTC11637 for an additional 6 h, and then harvested for further analysis. For the transfection experiment, the cells were co-transfected with 50 nM siRNA and 2 μg pcDNA3.1/cagA in the presence of 6 μl Lipofectamine 2000 for 48 h, and then harvested. The scraped siRNA served as control.
Transcription factor binding sites in the gastrin promoter
Transcription factor binding sites in the gastrin promoter were analyzed with AliBaba2.1 (http://www.gene-regulation.com) and TESS (http://www.cbil.upenn.edu/cgi-bin/tess).
Statistical analysis
Results are expressed as means ± SEM. Statistical treatment was performed with SPSS12.0 software (SPSS, Chicago, IL, USA). One-way analysis of variance (ANOVA) was used to analyze the data. Statistical significance was established at P < 0.05.
Results
Characteristics of the expression vector pcDNA3.1/cagA and luciferase reporter vector pGL/GP
To study the relationship between CagA and gastrin expression, we constructed a eukaryotic expression vector containing the cagA gene, pcDNA3.1/cagA, and a luciferase reporter vector harboring the gastrin promoter, pGL/GP. The two vectors were identified through sequencing. The full-length of the cagA sequence was 3467 bp and the length of the gastrin promoter was 1347 bp located upstream from the first exon of gastrin mRNA. The two sequences have been submitted to GenBank and have accession numbers GQ161098 and HM151000, respectively.
Determining CagA in transfected and infected gastric cancer cells
After the expression vector pcDNA3.1/cagA was successfully constructed, we transfected it into gastric cancer AGS and SGC-7901 cells. Alternatively, the two cells were infected with cagA-positive H. pylori NCTC11637. As shown in Fig. 1a, b, CagA was present in the transfected and infected cells as well as H. pylori NCTC11637. However, the phosphorylated CagA was only present in the infected cells and H. pylori NCTC11637, but was absent in the transfected cells (data not shown).

Expression of CagA, phosphorylated CagA, and gastrin. a cagA mRNA (top row) and protein (third row down) expression is measured by quantitative reverse transcription polymerase chain reaction (RT-PCR) and immunoblotting in AGS and SGC-7901 cells transfected with pcDNA3.1/cagA. b CagA and phosphorylated CagA proteins are measured by immunoblotting in AGS and SGC-7901 cells infected with Helicobacter pylori NCTC11637. c Gastrin mRNA expression is measured by RT-PCR in AGS and SGC-7901 cells. Hp, H. pylori NCTC11637; P-CagA, phosphorylated CagA; pcDNA3.1/cagA, vector containing cagA gene. The data are the average of three independent experiments. The gels and hyperfilms are cut to make clearer and more concise figures. GAPDH Glyceraldehyde-3-phosphate-dehydrogenase
Up-regulation of gastrin mRNA expression by CagA in gastric cancer cells
After the presence of CagA in the infected and transfected AGS and SGC-7901 cells was confirmed, we transfected the pGL/GP vector into the AGS and SGC-7901 cells. As shown in Fig. 2a, b, the luciferase activity was significantly increased in both the infected and transfected cells, suggesting that CagA can activate the gastrin promoter.

CagA enhances gastrin promoter activity and gastrin mRNA level. Luciferase activity is measured with the Dual-Luciferase Reporter Assay System in either Hp-infected (a) or cagA-transfected (b) AGS and SGC-7901 cells. The luciferase activity is normalized to Renilla luciferase and the results are expressed as a percentage relative to the control. Gastrin mRNA expression is measured by quantitative RT-PCR in either Hp-infected (c) or cagA-transfected (d) AGS and SGC-7901 cells. Gastrin mRNA level is normalized to β-actin by a comparative Ct method and the results are expressed as percentages relative to the control. Hp, H. pylori NCTC11637; D-Hp, Hp boiled for 15 min; pcDNA3.1/cagA, vector containing cagA gene; pGL/GP, vector containing gastrin promoter. *P < 0.05 obtained by analysis of variance (ANOVA) for comparison with Lane 2. The data shown are the average of three independent experiments
Next, we wondered if CagA could up-regulate gastrin gene expression? The results showed that after the AGS and SGC-7901 cells, which constitutively express gastrin (Fig. 1c), were infected with H. pylori NCTC11637 or transfected with the pcDNA3.1/cagA, the gastrin mRNA level was increased (Fig. 2c, d), and both the relative luciferase activity and gastrin mRNA level were much higher in the transfected cells than in the infected cells, suggesting that CagA protein may be a very important factor in the induction of gastrin expression.
Effect of MEK/ERK-signaling inhibition on CagA-induced gastrin expression in gastric cancer cells
To elucidate that the signaling pathway(s) mediated CagA-induced gastrin expression, we first confirmed the role of MEK/ERK-dependent signaling in this induction, because ERK kinase can be activated by CagA translocated into gastric epithelial cells after infection with H. pylori. In this study, after pretreatment with the MEK inhibitor U0126 (upstream activator of ERK), gastrin promoter activity and gastrin mRNA expression induced by H. pylori infection were significantly inhibited (Fig. 3a, c). Similar results were also observed in cagA-transfected cells, and the inhibitory effect of U0126 was stronger in these cells (Fig. 3b, d).

U0126 inhibits CagA-induced gastrin promoter activity and gastrin mRNA expression. Luciferase activity is measured with the Dual-Luciferase Reporter Assay in AGS and SGC-7901 cells pretreated with U0126 and either infected with H. pylori (a) or transfected with pcDNA3.1/cagA (b). The luciferase activity is normalized to Renilla luciferase and the results are expressed as percentages relative to the control. Gastrin mRNA expression is measured by quantitative RT-PCR in AGS and SGC-7901 cells pretreated with U0126 and either infected with H. pylori (c) or transfected with pcDNA3.1/cagA (d). Gastrin mRNA level is normalized to β-actin by a comparative Ct method and the results are expressed as percentages relative to the control. Hp, H. pylori NCTC11637; D-Hp, Hp boiled for 15 min; U0126, MEK/ERK signaling pathway inhibitor; pcDNA3.1/cagA, vector containing cagA gene; pGL/GP, vector containing gastrin promoter. *P < 0.05 obtained by ANOVA for comparison with Lane 2. The data shown are the average of three independent experiments
Next, we knocked-down ERK1 expression with ERK1-siRNA, and found that the knockdown of endogenous ERK1 expression resulted in the inhibition of CagA-induced gastrin mRNA expression in H. pylori-infected and cagA-transfected gastric cancer cells (Fig. 4).

Knockdown of ERK1 inhibits CagA-induced gastrin mRNA expression. a Gastrin mRNA expression is measured by quantitative RT-PCR in both Hp-infected and ERK1-siRNA transfected AGS and SGC-7901 cells. b Gastrin mRNA expression is measured by quantitative RT-PCR in AGS and SGC-7901 cells co-transfected with both pcDNA3.1/cagA and ERK1-siRNA. Gastrin mRNA level is normalized to β-actin by a comparative Ct method and the results are expressed as percentages relative to the control. c ERK1/2 protein expression is measured by immunoblotting in AGS and SGC-7901 cells transfected with ERK1-siRNA. Hp, H. pylori NCTC11637; D-Hp, Hp boiled for 15 min; pcDNA3.1/cagA, vector containing cagA gene; siRNA, small interfering RNA. *P < 0.05 obtained by ANOVA for comparison with Lane 3. The data shown are the average of three independent experiments
Involvement of JAK2-dependent signaling in CagA-induced gastrin expression
Previous studies have shown that, besides MEK/ERK signaling, CagA can also activate the JAK/STAT (STAT: Signal transducers and activators of transcription) pathway in vivo and in vitro. To find whether JAK-signaling participated in CagA-induced gastrin expression, the JAK2 inhibitor AG490 and JAK2-siRNA were used to treat the H. pylori-infected and cagA-transfected gastric cancer cells, and this resulted in the significant inhibition of luciferase activity and gastrin mRNA expression in the cells (Fig. 5). Similar to the blocking of MEK/ERK signaling, the inhibitory effects of AG490 and JAK2-siRNA were much greater in cagA-transfected cells than in H. pylori-infected cells.

AG490 and JAK2-siRNA inhibit gastrin promoter activity and mRNA expression. Luciferase activity is measured with the Dual-Luciferase Reporter Assay System in AGS and SGC-7901 cells pretreated with AG490 and either infected with H. pylori (a) or transfected with pcDNA3.1/cagA (b). The luciferase activity is normalized to Renilla luciferase and the results are expressed as percentages relative to the control. Gastrin mRNA expression is measured by quantitative RT-PCR in AGS and SGC-7901 cells pretreated with AG490 and either infected with H. pylori (c) or transfected with pcDNA3.1/cagA (d). Gastrin mRNA level is normalized to β-actin by a comparative Ct method and the results are expressed as percentages relative to the control. e JAK2 protein expression is measured by immunoblotting in AGS and SGC-7901 cells transfected with JAK2-siRNA. Hp, H. pylori NCTC11637; D-Hp, Hp boiled for 15 min; AG490, JAK2 signaling pathway inhibitor; pcDNA3.1/cagA, vector containing cagA gene; pGL/GP, vector containing gastrin promoter. *P < 0.05 obtained by ANOVA for comparison with Lane 2. # P < 0.05 obtained by ANOVA for comparison with Lane 4. The data shown are the average of three independent experiments
Transcription factor binding sites in gastrin promoter
AliBaba2.1 and TESS analysis indicated that there were 3 activator protein 1 (AP1), one cyclic adenosine monophosphate [cAMP] responsive element binding protein (CREB), 9 nuclear factor I (NF I), one nuclear factor kappa B NFκB, 38 sequence-specific transcription factor 1 (SP1), and 4 STAT binding sites in the gastrin promoter.
Discussion
Gastrin, a small-molecule polypeptide (molecular weight <4 kDa), has been proven to cause stomach cancer [9]. Hur et al. [10] found that the expression level of gastrin was high in gastric carcinoma tissues; and in patients with diffuse-type gastric carcinoma, the patients with positive gastrin had a poorer prognosis than the patients with negative gastrin, suggesting that gastrin is an autocrine growth factor in diffuse-type gastric carcinoma. Gastrin was also proven to be an independent predictor of gastric carcinoma in a multivariate analysis [11]. A recent report has indicated that the great majority of cells which express CD133 and CD44 also express gastrin precursors in human colorectal cancer samples and stem cell-like DLD-1 cell lines, suggesting that there may be gastrin expression in tumor stem cells, and that gastrin is associated with the invasion and metastases of gastrointestinal cancer [12]. Exogenous gastrin-17 can also promote the migration of human gastric cancer cells through the mixed lineage kinase-3/JNK1 axis [13]. These data demonstrate that gastrin is a crucial factor in the development and progression of gastric cancer.
H. pylori infection is an important independent risk factor for gastric carcinogenesis. It has been confirmed that H. pylori infection can result in hypergastrinemia [14]. Asymptomatic patients with H. pylori colonization have elevated serum gastrin concentrations [15], and following the eradication of H. pylori, there is a reduction of serum gastrin concentration in fasting patients [16]. In 2009, Takaishi et al. [17] found a marked effect of gastrin on carcinogenesis of both the gastric corpus and the antrum in mice with H. pylori infection, suggesting that gastrin and H. pylori are essential cofactors for carcinogenesis of the gastric corpus. However, the correlation between H. pylori infection and gastrin expression in gastric carcinogenesis has been unclear.
Among the virulence factors of H. pylori, CagA protein is a crucially carcinogenic factor of gastric cancer. After infection with cagA-positive H. pylori, CagA protein is directly delivered into gastric epithelial cells via the type IV secretion system [10]. We also detected the presence of CagA protein in gastric cancer AGS and SGC-7901 cells infected with H. pylori NCTC11637 (Fig. 1b).
Subsequently, we explored the possibility that the gastrin promoter is a transcriptional target of CagA in gastric cancer cells. We observed that either H. pylori infection or forced cagA expression significantly enhanced the transcription activity of the gastrin promoter and gastrin mRNA levels in gastric cancer cells (Fig. 2), suggesting that CagA can up-regulate gastrin gene expression through activating the gastrin promoter. Furthermore, in our studies, both the relative luciferase activity and gastrin mRNA level in cagA-transfected cells were much higher than these values in H. pylori-infected cells (Fig. 2), further demonstrating that CagA plays a crucial role in inducing gastrin expression. Similarly, CagA protein expression in the transfected cells, especially in AGS cells, was higher than that in the infected cells (Fig. 1a, b).
Some researchers believe that after being translocated into gastric epithelial cells, CagA is phosphorylated on several specific tyrosine sites, EPIYA (Glu-Pro-Ile-Tyr-Ala) sites, by Src family and Abl kinases, which may be an important mechanism of gastric carcinogenesis [18]. However, other studies have confirmed that the translocated CagA plays a pathogenic role in a tyrosine-phosphorylated-independent manner. Our results showed that the tyrosine-phosphorylated CagA was present in H. pylori NCTC 11637 without having any contact with host cells and there was no significant difference between the levels of CagA and tyrosine-phosphorylated CagA in either NCTC11637 or infected cells (Fig. 1b). The tyrosine-phosphorylated CagA in the cells which transiently expressed CagA was not able to be detected (data not shown). These data suggest that CagA may up-regulate gastrin mRNA expression in a tyrosine-phosphorylation-independent manner. Recently, Suzuki et al. [19] have reported that the C-terminal region activity of CagA leads to epithelial proliferation and the proinflammatory response is associated with the development of chronic gastritis and gastric cancer.
The translocated CagA can interact with several proteins in host cells, especially SHP-2 phosphatase, resulting in the activation of several signaling pathways, including the ERK/MAPK and JAK/STAT pathways [20]. SHP-2 can activate MEK/ERK and MAPK kinases by both Ras-dependent and Ras-independent mechanisms [21]. Higashi et al. [22] have also found that ERK is activated by CagA via SHP-2 recruitment and activation in AGS cells. Similarly, CagA expression in gastric epithelial cells results in sustained ERK MAP kinase activation [23]. The research of Zhu et al. [24] further confirms that the transformation of immortalized gastric epithelial cells is induced by CagA via the ERK/MAPK pathway. Activated ERK protein enters the cell nucleus to activate important tumor-related transcription factors such as AP1, CREB, NF I, NFκB, SP1, and myelocytomatosis viral oncogene homolog (MYC) [25]. Our analysis indicated that there were 3 AP1, one CREB, 9 NF I, one NFκB, 38 SP1, and 4 STAT binding sites in the gastrin promoter. These binding sites can mediate the combination of ERK with the gastrin promoter.
Besides the MEK/ERK signaling pathway, CagA can also activate the STAT3 signaling pathway in vitro and in vivo, providing a potential mechanism by which chronic H. pylori infection promotes the development of gastric cancer [26]. The SHP-2 phosphatase can also induce the activation of the JAK/STAT signaling pathway [27]. The gastrin promoter also has 4 STAT binding sites. We also found that the activation of the MEK/ERK and JAK/STAT signaling pathways was likely to be associated with CagA-induced high expression of gastrin in gastric cancer cells, because the two pathway inhibitors, U0126 and AG490, and ERK1 and JAK2-siRNA all significantly inhibited CagA-induced gastrin promoter activation and gastrin mRNA expression (Figs. 3, 4, 5), and their inhibitory effects were more powerful in the cagA-transfected cells.
Gastric cancer is a common malignant gastrointestinal tumor in Asia. It comes second in global cancer mortality. H. pylori infection is the initial factor of gastric cancer. Based on our experimental results, we speculate that after gastric epithelial cells are infected by H. pylori, CagA starts the MEK/ERK and JAK/STAT signaling pathways to activate tumor-related transcription factors such as AP1, CREB, NFκB, and STAT, which combine with their corresponding binding sites in the gastrin promoter, leading to the up-regulation of gastrin mRNA expression. Gastrin combines with gastrin receptors and forms an autocrine loop which stimulates cellular proliferation, resulting in stomach carcinogenesis. This may be a main cause of stomach carcinogenesis and may become a new treatment target in gastric cancer.
In summary, our study suggests that the gastrin promoter may be the transcriptional target of CagA, and that CagA can activate the gastrin promoter to up-regulate gastrin mRNA expression in gastric cancer cells. The activation of the MEK/ERK and JAK signaling pathways may be involved in this mechanism by the connection of binding elements for ERK targets with STAT binding sites in the gastrin promoter. Our study will help to further explore the mediator in the development of gastric cancer caused by H. pylori infection and the search for treatment targets of gastric cancer.
References
Ferrand A, Wang TC. Gastrin and cancer: a review. Cancer Lett. 2006;238:15–29.
Sun FP, Song YG. G and D Cells in rat antral mucosa: an immunoelectron microscopic study. World J Gastroenterol. 2003;9:2768–71.
Tu S, Chi AL, Lin S, Dubeykovskaya Z, Ai W, Fleming JV, et al. Gastrin regulates the TFF2 promoter through gastrin-responsive cis-acting elements and multiple signaling pathways. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1726–37.
Hatakeyama M. The role of Helicobacter pylori CagA in gastric carcinogenesis. Int J Hematol. 2006;84:301–8.
Konturek SJ, Stauaynska T, Konturek PC, Karczewska E, Marlicz K, Lawniczak M, et al. Helicobacter pylori and CagA status, serum gastrin, interleukin-8 and gastric acid secretion in gastric cancer. Scand J Gastroenterol. 2002;37:891–8.
Konturek PC, Hartwich A, Zuchowicz M, Labza H, Pierzchalski P, Karczewska E. Helicobacter pylori, gastrin and cyclooxygenases in gastric cancer. J Physiol Pharmacol. 2000;51:737–49.
Ding SZ, Olekhnovich IN, Cover TL, Peek RM Jr, Smith M, et al. Helicobacter pylori and mitogen- activated protein kinases mediate activator protein-1 (AP-1) subcomponent protein expression and DNA-binding activity in gastric epithelial cells. FEMS Immunol Med Microbiol. 2008;53:385–94.
Jan H, Schefe·Kerstin E, Lehmann IR, Unger T, Funke-Kaiser H. Quantitative real-time RT-PCR data analysis: current concepts and the novel “gene expression’s CT difference” formula. J Mol Med. 2006;84:901–10.
Ito M, Tanaka S, Maeda M, Takamura A, Tatsugami M, Wada Y, et al. Role of the gastrin-gastrin receptor system in the expansive growth of human gastric neoplasms. Digestion. 2008;78:163–70.
Hur K, Kwak MK, Lee HJ, Park DJ, Lee HK, Lee HS, et al. Expression of gastrin and its receptor in human gastric cancer tissues. J Cancer Res Clin Oncol. 2006;132:85–91.
Stephens MR, Hopper AN, Lewis WG, Blackshaw G, Edwards P, Osborne B, et al. Prognostic significance of gastrin expression in patients undergoing R0 gastrectomy for adenocarcinoma. Gastric Cancer. 2007;10:159–66.
Ferrand A, Sandrin MS, Shulkes A, Baldwin GS. Expression of gastrin precursors by CD133-positive colorectal cancer cells is crucial for tumor growth. Biochim Biophys Acta. 2009;1793:477–88.
Mishra P, Senthivinayagam S, Tangasamy V, Sondarva G, Rana B. Mixed lineage kinase-3/JNK1 axis promotes migration of human gastric cancer cells following gastrin stimulation. Mol Endocrinol. 2010;24:598–607.
Fossmark R, Qvigstad G, Waldum HL. Animal studies on the risk of hypoacidity and hypergastrinemia. World J Gastroenterol. 2008;14:1646–51.
Smith JT, Pounder RE, Nwokolo CU, Lanzon-Miller S, Evans DG, Graham DY, et al. Inappropriate hypergastrinaemia in asymptomatic healthy subjects infected with Helicobacter pylori. Gut. 1990;31:522–5.
Levi S, Beardshall K, Swift I, Foulkes W, Playford R, Ghosh P, et al. Antral Helicobacter pylori, hypergastrinaemia, and duodenal ulcers: effect of eradicating the organism. BMJ. 1989;299:1504–5.
Takaishi S, Tu S, Dubeykovskaya ZA, Whary MT, Muthupalani S, Rickman BH, et al. Gastrin is an essential cofactor for helicobacter-associated gastric corpus carcinogenesis in C57BL/6 mice. Am J Pathol. 2009;175:365–75.
Hatakeyama M. Helicobacter pylori and gastric carcinogenesis. J Gastroenterol. 2009;44:239–48.
Suzuki M, Mimuro H, Kiga K, Fukumatsu M, Ishijima N, Morikawa H, et al. Helicobacter pylori CagA phosphorylation-independent function in epithelial proliferation and inflammation. Cell Host Microbe. 2009;5:23–34.
Lai YP, Yang JC, Lin TZ, Lin JT, Wang JT. Helicobacter pylori infection and CagA protein translocation in human primary gastric epithelial cell culture. Helicobacter. 2006;11:451–9.
Schaeper U, Gehring NH, Fuchs KP, Sachs M, Kempkes B, Birchmeier W. Coupling of Gab1 to c-Met, Grb2, and Shp2 mediates biological responses. J Cell Biol. 2000;149:1419–32.
Higashi H, Nakaya A, Tsutsumi R, Yokoyama K, Fujii Y, Ishikawa S, et al. Helicobacter pylori CagA induces Ras-independent morphogenetic response through SHP-2 recruitment and activation. J. Biol Chem. 2004;279:17205–16.
Higuchi M, Tautsumi R, Higashi H, Hatakeyama M. Conditional gene silencing utilizing the lac repressor reveals a role of SHP-2 in cagA-positive Helicobacter pylori pathogenicity. Cancer. 2004;95:442–7.
Zhu Y, Zhong X, Zheng S, Du Q, Xu W. Transformed immortalized gastric epithelial cells by virulence factor CagA of Helicobacter pylori through Erk mitogen-activated protein kinase pathway. Oncogene. 2005;24:3886–95.
Mebratu Y, Tesfaigzi Y. How ERK1/2 activation controls cell proliferation and cell death. Is subcellular localization the answer? Cell Cycle. 2009;8:1168–75.
Bronte-Tinkew DM, Terebiznik M, Franco A, Du Q, Xu W. Helicobacter pylori cytotoxin-associated gene A activates the signal transducer and activator of transcription 3 pathway in vitro and in vivo. Cancer Res. 2009;69:632–9.
Yu WM, Hawley TS, Hawley RG, Qu CK. Catalytic-dependent and -independent roles of SHP-2 tyrosine phosphatase in interleukin-3 signaling. Oncogene. 2003;22:5995–6004.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (No. 30560038), the Project of International Cooperation in Science and Technology of Guizhou Province (NO, G(2008) 700119), and the Science and Technology Foundation of GuiZhou Province (NO. [2007] 2084). We thank the Chinese Center of Helicobacter pylori strain Management and Preservation for providing H. pylori NCTC11637. We also thank Dr Yang WenXiu and Dr Wu JiaHong for technical assistance.
Conflict of interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
J. Zhou and Y. Xie contributed equally to this work.
Rights and permissions
About this article
Cite this article
Zhou, J., Xie, Y., Zhao, Y. et al. Human gastrin mRNA expression up-regulated by Helicobacter pylori CagA through MEK/ERK and JAK2-signaling pathways in gastric cancer cells. Gastric Cancer 14, 322–331 (2011). https://doi.org/10.1007/s10120-011-0044-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10120-011-0044-2