
Abstract
Background
Bevacizumab is approved for various cancers. This analysis aimed to comprehensively evaluate bevacizumab pharmacokinetics and the influence of patient variables on bevacizumab pharmacokinetics.
Methods
Rich and sparse bevacizumab serum concentrations were collected from Phase I through IV studies in early and metastatic cancers. Bevacizumab was given intravenously as single agent or in combination with chemotherapy for single- and multiple-dose schedules.
Results
Model-building used 8943 bevacizumab concentrations from 1792 patients with colon/colorectal, non-small cell lung, kidney, pancreatic, breast, prostate and brain cancer. Bevacizumab doses ranged from 1 to 20 mg/kg given once every 1, 2 or 3 weeks. A two-compartment model best described the data. The population estimates of clearance (CL), central volume of distribution (V1) and half-life for a typical 70-kg patient were 9.01 mL/h, 2.88 L and 19.6 days. CL and V1 increased with body weight and were higher in males than females by 14 and 18 %, respectively. CL decreased with increasing albumin and decreasing alkaline phosphatase. The final model was externally validated using 1670 concentrations from 146 Japanese patients that were not used for model-building. Mean prediction errors were −2.1, 3.1 and 1.0 % for concentrations, CL and V1, respectively, confirming adequate predictive performance.
Conclusions
A robust bevacizumab pharmacokinetic model was developed and externally validated, which may be used to simulate bevacizumab exposure to optimize dosing strategies. Asian and non-Asian patients exhibited similar bevacizumab pharmacokinetics. Given the similarity in pharmacokinetics among monoclonal antibodies, this may inform pharmacokinetic studies in different ethnic groups for other therapeutic antibodies.
Similar content being viewed by others
Introduction
Bevacizumab (Avastin®, Genentech Inc.) is a humanized monoclonal immunoglobulin G (IgG) 1 antibody that specifically binds and neutralizes the biological activity of vascular endothelial growth factor A (VEGF-A), a key isoform of VEGF involved in angiogenesis, and a well-characterized pro-angiogenic factor [1]. Bevacizumab causes inhibition of tumor angiogenesis by blocking VEGF-A from binding to its receptors and leads to tumor growth inhibition. Bevacizumab in combination with standard therapy has received marketing authorization for use in the treatment of various cancers including metastatic colorectal cancer (CRC) [2, 3], non-small cell lung cancer (NSCLC) [4], breast cancer [5], renal cell carcinoma [6], cervical cancer [7] and ovarian cancer [8].
A population pharmacokinetic (PK) model has been previously developed [9]. Bevacizumab PK showed dose linearity within the dose range of 1–20 mg/kg, a slow clearance, a volume of distribution consistent with limited extravascular distribution and a terminal half-life of approximately 20 days. Clearance (CL) and central volume of distribution (V1) increased with body weight and were higher in male patients. CL decreased with increasing albumin and decreasing alkaline phosphatase. There has been no evidence for anti-therapeutic antibodies (ATAs) for bevacizumab in metastatic solid tumors based on the large number of historical clinical studies, and ATA was detected in only 0.6 % of the patients with colon cancer (adjuvant setting) [10].
However, the previous analysis had several limitations. Several important covariates were not evaluated in previous analysis including ethnicity (e.g., Asian vs. non-Asian), indications and baseline VEGF-A. First, bevacizumab has been widely used across ethnic groups (e.g., Asian vs. non-Asian), and supplementary Biologics License Applications have been submitted to health authorities for approval of using bevacizumab for new indications or new combinations based on data from limited ethnic groups while the target population contains much broader ethnic groups. Therefore, it is important to evaluate ethnicity (Asian vs. non-Asian) as a covariate. Second, it has been shown that bevacizumab clearance is 50 % higher and exposure is 50 % lower in gastric cancer as compared to other types of solid tumors [11], making it important to evaluate indication as a covariate. Finally, several studies have shown the predictive value of baseline VEGF-A for bevacizumab treatment effect on progression-free survival and/or overall survival, meaning that only patients with high VEGF-A levels may benefit from bevacizumab treatment, for example in gastric cancer [12] and metastatic breast cancer [13]. Therefore, it is important to evaluate baseline VEGF-A as a covariate. The reason why these important covariates were not evaluated in the previous analysis is likely that these evidences showing the importance of these covariates all appeared after the previous analysis, and therefore, the significance of these covariates may not have been fully realized at that time, and/or the data were unavailable at that time.
Other limitations of the previous analysis include utilization of FO (first-order) instead of FOCE (first-order conditional estimation) algorithm in NONMEM [14], limited number of studies (n = 6), patients (n = 491) and indications (mainly CRC, NSCLC and breast cancer), etc. Therefore, an updated analysis is warranted.
The objectives of the current analysis were to develop a robust population PK model in adult patients with solid tumors and to evaluate the influence of patient variables on bevacizumab PK, which can be used to simulate bevacizumab exposure to optimize bevacizumab dosing strategies.
Methods
Patients
Studies in adult cancer patients included in this analysis are summarized in Table 1. Patients with at least one PK sample were evaluated. Serum bevacizumab concentrations were determined at Genentech, Inc., using an enzyme-linked immunosorbent assay that used recombinant human VEGF for capture and a goat antibody to human IgG conjugated to horseradish peroxidase for detection. The lowest limit of quantification (LLOQ) was 78 ng/mL in serum [9]. Concentrations below the LLOQ were omitted. The clinically relevant covariates tested included those related to demographics, biochemical tests, concomitant medications and pathophysiological factors (Table 2). Values of covariates that follow lognormal distribution were log-transformed. Bevacizumab was given via intravenous infusion of 30–90 min in all patients.
Population pharmacokinetic modeling
A population PK model was developed using data from 15 studies: AVF0737g, AVF0757g, AVF0761g, AVF0775g, AVF0776g, AVF0780g, AVF2107g, AVF2119g, AVF3077s, BO17704, BO17705, BO17706, BP20689, BO21015 and BO21990. Nonlinear mixed-effects modeling was performed with NONMEM (version 7.2; ICON Development Solutions, Ellicott City, Maryland, USA) [14] using the FOCE method with interaction, Perl-speaks-NONMEM (version 3.5.3; Uppsala University, Uppsala, Sweden) [15] and R 3.0.3 [16]. Several models with various residual error structures and OMEGA matrices were tested to select the optimal base model. The base model included a power function of body size [e.g., total body weight (BWT)] on all PK parameters:
where BWT i = baseline BWT of patient i; P i = typical PK parameters of patients with BWT i ; P TV = typical value of PK parameters for patients with BWT of 70 kg; θ P = exponent for the PK parameter P.
The base model was evaluated using either theoretical (0.75 for clearances and 1 for volumes of distribution) [17] or fitted values of exponents θ P . The quality of fit was evaluated using a standard model discrimination process including statistical criteria [i.e., minimum of objective function value (OFV)], adequate estimation of the parameters (e.g., relative standard error <50 %) and graphical representations of goodness of fit. The final model was established in a stepwise manner by forward addition followed by backward elimination of parameter–covariate relationships with a significance level of p < 0.01 and p < 0.001, respectively (OFV decrease of 6.63 and 10.83 for one degree of freedom based on Chi-squared distribution, respectively).
The effect of n covariates at baseline on PK parameters was coded using a multiplicative model:
where θ i is the typical value of the parameter for patients with a set of covariates i, θ TV is the typical value of the PK parameter for patients having the covariate values equal to the median of the covariate for all patients, and Effect1,i through Effect n,i are multiplicative factors of the effects for covariate 1 through n, for the set of covariates i. The covariate models for both continuous and categorical covariates were chosen to avoid prediction of negative parameter values.
The multiplicative factor was defined using the power function for continuous covariates:
and defined as follows for categorical covariates:
-
if this categorical covariate is equal to 0, then Effect i = 1
-
if this categorical covariate is not equal to 0, then Effect i = eθeff
where Effect i is the multiplicative factor of the covariate effect for covariate i at baseline, Cov i is the covariate value, Covreference value is the median of the covariate for all patients, and θ eff is the exponent of the power function to be estimated. eθeff was used for categorical covariates to force a positive value.
For patients with missing value for a continuous covariate Cov i , the multiplicative factor of Cov i was calculated as [18]:
where θ cov missing is the value of the covariate that was estimated by fitting to the data from all patients with missing information. During the forward addition step, θ cov missing was estimated by fitting to the data for all covariates with missing values. During the backward elimination step, θcov missing was fixed to the estimates obtained from the forward addition step to stabilize the model. All patients with missing value had the same estimate of θ cov missing.
Model evaluation and sensitivity analysis
The population PK models were evaluated using diagnostic plots [19, 20], visual predictive check (VPC) [20, 21], prediction-corrected VPC (pcVPC) [22], bootstrapping [23] and shrinkage [24] assessments. VPC, pcVPC and bootstrapping were all performed using 2000 replicates based on the final model. The relative impact of each covariate included in the final model alone on PK parameters and exposure was explored. Exposure including the trough (C min) and peak (C max) concentration was computed at steady state given bevacizumab of 10 mg/kg once every 2 weeks using the final model. The computation was performed using the extreme covariate values (5th and 95th percentiles) and the equations of the final model.
External validation
After the final model was built, data from three Japanese studies (JO18157, JO19901 and JO19907, Tables 1 and 2) became available and were subsequently used for external validation. Predicted bevacizumab concentrations (C IPRED) for the validation population were obtained using post hoc Bayesian forecasting by fixing the parameters in the structural and variance models to the final estimates. Prediction errors (PEs) as a measure of bias were calculated for each concentration as PE = (C IPRED − C OBS)/C OBS, where C OBS denotes observed concentrations. Root mean squared prediction error (RMSE) as a measure of precision was calculated as \( \sqrt {\frac{1}{n}\sum (C_{\text{IPRED}} - C_{\text{OBS}} )^{2} } \), where n denotes the number of observations. pcVPC approach was used to compare the 95 % prediction interval and OBS.
Predicted PK parameters (P IPRED) for each patient were calculated based on individual covariate values using the equations in the final model without considering observed concentrations. Post hoc estimates of PK parameters (P EST) were obtained based on observed concentrations and the final model. PE was calculated as (P IPRED − P EST)/P EST. RMSE was calculated as \( \sqrt {\frac{1}{n}\sum (P_{\text{IPRED}} - P_{\text{EST}} )^{2} } \), where n denotes the number of patients.
Results
Patients
A total of 8943 bevacizumab serum concentrations from 1792 adult cancer patients in 15 studies were included in the model-building dataset, and 1670 concentrations from 146 adult patients in three Japanese studies were included in the external validation dataset. Studies and patient characteristics are summarized in Tables 1 and 2. Less than 5 % of the samples were below LLOQ, and all of them were pre-dose samples.
Population pharmacokinetic modeling
The optimal base model was a linear two-compartment model with theoretical exponents estimated for clearance (CL), inter-compartment clearance (Q), central (V1) and peripheral (V2) volumes of distribution, full block inter-individual variability (IIV) on CL, V1 and V2 with combined additive and proportional residual error. Parameter estimates of the base model are presented in Supplementary Table 1. In the base model, the estimates of typical bevacizumab CL, V1, Q, V2 and terminal half-life values for a 70-kg patient were 9.01 mL/h, 2880 mL, 18.7 mL/h, 2571 mL and 19.6 days. Thirty-eight covariate relationships were evaluated in the forward addition step. After adjusting for total body weight (BWT), CL and V1 were still higher in male patients. CL decreased with increasing albumin (ALBU) and decreasing baseline alkaline phosphatase (BALP). CL was also lower in patients treated with interferon alpha (Supplementary Fig. 1). No covariate was excluded during the backward elimination step (p < 0.001).
Parameter estimates of the final model are summarized in Table 3. Bevacizumab CL and V1 for the patient i were described as follows (ALBU = 41.8 and BALP = 76.3 if missing):
Model evaluation and sensitivity analysis
Goodness-of-fit plots showed good agreement between predicted and observed bevacizumab concentrations with no apparent bias in residual (Supplementary Fig. 2). The pcVPC result for the final model is presented in Fig. 1. Overall, the 2.5th, 50th and 97.5th percentiles of observed concentrations were within the predicted 95 % confidence interval (CI) of these percentiles, suggesting accurate model fitting across a wide range of dosing regimens and time courses. Bootstrapping resulted in median parameter estimates and 95 % CIs similar to the estimates from the original dataset, indicating that the final model provided good precision for parameter estimation.

Prediction-corrected visual predictive check for the serum concentration-time profiles of bevacizumab using the final model in adult cancer patients. Pred population prediction; figure on the right is the part of figure on the left during the first 2 months after dose
The impact of the variation for a single covariate included in the final model on steady-state exposure (Fig. 2a, b), CL (Fig. 2c) and V1 (Fig. 2d) and steady-state exposure (Fig. 2) is demonstrated by comparing the simulated CL, V1 and exposure of patients with extreme covariate values (5th and 95th percentiles) to a typical patient with median covariate value. Among all covariates, BWT had the strongest impact on CL (change at extreme BWT values: −17.4 to 30.3 %) and V1 (change at extreme BWT values: −14.1 to 23.5 %). The impact of other covariates on CL (<22 %) and V1 (<18 %) was low. BWT had the strongest impact on C min (30.3 %) and C max (37.6 %). The impact of the variation for other covariates on C min and C max was all below 30 %.


Impact of the variation for a single covariate included in the final model on steady-state bevacizumab exposure and PK parameters in adult cancer patients: a C min (minimum concentration); b C max (maximum concentration); c CL (clearance); d V1 (central volume of distribution). Red vertical lines represent the “base” defined as the exposure or PK parameter estimate of a typical patient, i.e., a 70-kg female patient with albumin of 39 g/L and baseline alkaline phosphatase of 109 U/L without interferon alpha treatment. The dark blue shaded curve at the bottom with value at each end shows the 5th to 95th percentile range of exposure or PK parameter estimate across the entire population. Each light blue shaded bar represents the influence of a single covariate on the steady-state exposure after repeated bevacizumab dose of 10 mg/kg once every 2 weeks or on the PK parameter. The label at left end of the bar represents the covariate being evaluated. The upper and lower values for each covariate capture 90 % of the plausible range in the population. The length of each bar describes the potential impact of that particular covariate on bevacizumab steady-state exposure or PK parameters, with the percentage value in the parentheses at each end representing the percent change from the “base.” The most influential covariate is at the bottom of the plot for each exposure metric or PK parameter
External validation
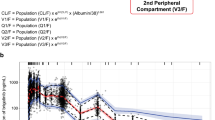
Over 95 % of prediction-corrected observations fell within the 95 % prediction interval (PI) (Fig. 3). CL and V1 calculated based on the equations in the final model (P IPRED) were similar to those estimated based on observed concentrations (P EST) (Fig. 4a, b). Mean PE for bevacizumab serum concentrations, CL and V1 were −2.1, 3.1 and 1.0 %, respectively. No bias in PE was observed over time and across predicted values. RMSE for bevacizumab serum concentrations, CL and V1 were 0.283, 0.017 and 2.60, respectively.

External validation. Most of prediction-corrected observations fall between the 95 % prediction intervals. There is no apparent systematic bias in prediction

Comparison between (a, b) individual CL (a) and V1 (b) calculated based on individual covariate values using the equations in the final model without considering observed concentrations and post hoc estimates of CL and V1 obtained based on observed concentrations and the final model in the external validation population, and between (c, d) post hoc Bayesian estimates of CL (c) and V1 (d) of the model-building population and external validation population after normalization by individual covariate values that were included in the final model. Gray diamond in the boxplots represents the mean. CL clearance, Cov covariates included in the final model. V1 central volume of distribution. In Fig. 4c, d, data points with CL < 3 mL/h (n = 2) or V1 < 1500 mL (n = 1) are not displayed
Post hoc Bayesian estimates of CL and V1 in the model-building population (mostly non-Asian patients) and external validation population (Japanese patients only) were also similar after normalization by individual covariate values that were included in the final model (Fig. 4c, d). The normalization was done by dividing the post hoc Bayesian estimates of CL and V1 by the individual covariate values in the form that appeared in the equations of the final model.
Discussion
This analysis is a comprehensive PK evaluation of bevacizumab in adult cancer patients in Phases I–IV studies as a single agent or in combination with chemotherapy for both single- and multiple-dose administration with both rich and sparse bevacizumab serum concentration data. A robust population PK model was built based on a large PK population of 1792 patients from 15 studies and then externally validated using data from 146 Japanese patients in three independent studies. This model consolidated all bevacizumab PK data in one model, can timely support simulations and decision making when needed, can help develop consistent pharmacokinetic messages of bevacizumab for investigators and health authorities given that multiple PK models have been developed for bevacizumab and contained inconsistent messages, and can support future studies of bevacizumab in other indications. As mentioned in the “Introduction,” many important covariates that were not evaluated in the previous analysis (e.g., Asian vs. non-Asian, indications, baseline VEGF-A) were evaluated in this analysis.
Typical population PK parameter estimates were similar as previously published [9]. The low IIV of 29 and 18.3 % observed for CL and V1 was typical for antibody drugs [25]. The pcVPC demonstrated adequate fit and predictive performance of the final model (Fig. 1). The median prediction (blue band) may appear to be slightly below the median observation (blue line) beyond day 112, suggesting a possible tendency of under-prediction. However, this tendency is likely irrelevant given (1) the small degree of under-prediction, (2) the sparseness of data beyond day 112, (3) the good predictive performance for 2.5th and 97.5th percentiles (red) across all time points, as well as (4) the complexity and heterogeneity of the data. The external validation (Figs. 3, 4) demonstrated good predictive performance of the final model with no apparent systemic bias and the similarity in bevacizumab PK between Asian and non-Asian adult cancer patients. Although there may appear to be a tendency of over-prediction of the variability (Fig. 3), this tendency is likely irrelevant because the model was built based on more heterogeneous data (15 studies over a decade across various ethnic groups) while the validation data were more homogeneous (three studies over a few years in Asian patients only).
Factors significantly associated with bevacizumab PK were similar as previously published [9]: CL and V1 increased with BWT and were higher in males, and CL decreased with increasing albumin and decreasing BALP. It is well known that CL of other IgG antibodies is faster in patients with lower serum albumin levels [25], likely due to two reasons. First, the level of albumin correlates with disease status. Second, the recycling of albumin and IgG is both mediated by FcRn (neonatal Fc receptor) [25], and therefore, albumin levels may reflect the abundance and efficiency of FcRn. The effect of BALP on bevacizumab CL is likely because BALP is an indicator of disease burden, such as liver or bone metastases. CL was found to be 15.6 % lower in patients treated with interferon alpha. However, this effect was within the overall PK variability and therefore may be clinically irrelevant.
Similar to the previous analysis [9], tumor burden was not included in the final model in this analysis. Among solid tumors, tumor burden is usually an indicator of disease severity and health status. It is usually defined as the sum of longest diameters of target lesions under RECIST (Response Evaluation Criteria in Solid Tumors) criteria for systematic tumors and under other criteria for other tumors (e.g., brain tumors). Inclusion of tumor burden in bevacizumab PK model may not be crucial. First, tumor burden as an indicator of disease burden and health status could already be represented by albumin and BALP in the model. Second, tumor burden as a source of VEGF-A (target of bevacizumab) is irrelevant for bevacizumab PK because bevacizumab molar concentration is thousands of times higher than that of VEGF-A [10], and there has been no evidence of target-mediated drug disposition (TMDD) for bevacizumab [10]. Third, in previous analyses, tumor burden alone showed relatively low impact on bevacizumab exposure in the sensitivity analysis (similar to Fig. 2, data not published). Finally, the final model demonstrated adequate fitting and superior predictive performance without incorporating tumor burden.
On the other hand, three factors made it impossible to test baseline tumor burden as a covariate in this analysis. First, tumor response criteria were inconsistent across these 15 studies that were conducted across a time span of over a decade. Several different versions of RECIST and other criteria (e.g., Macdonald criteria for glioblastoma in BO21990) were used. Second, the methods used to measure tumor burden were inconsistent across studies, such as CT (computerized tomography) scans and MRI (magnetic resonance imaging). Finally, unit of length (mm) and area (mm2) both exist in tumor burden data, which cannot be converted to each other. In fact, inclusion of tumor burden in the model would greatly reduce the applicability of the model due to the continuous advancement in tumor response criteria and measurement methods, and due to different tumor response criteria and measurement methods across cancer types, for example RECIST version 1.0 versus version 1.1, RECIST criteria versus Macdonald criteria or RANO (Response Assessment in Neuro-Oncology) criteria, CT scans versus MRI.
In conclusion, a robust population PK model for bevacizumab in adult cancer patients was built and externally validated, which may be used to simulate concentration-time profile in adult cancer patients in future studies [11, 26]. Baseline body weight, albumin, alkaline phosphatase and gender were the covariates with the greatest influence on bevacizumab CL and V1, supporting body weight-based dosing of bevacizumab. No difference in bevacizumab PK was observed between Asian and non-Asian patients. Given the similarity in PK among many monoclonal antibodies, this may inform PK studies in different ethnic groups (e.g., Asian vs. non-Asian) for other therapeutic antibodies without TMDD and significant race-dependent target expression.
References
Ferrara N, Gerber HP, LeCouter J (2003) The biology of VEGF and its receptors. Nat Med 9:669–676
Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E, Ferrara N, Fyfe G, Rogers B, Ross R, Kabbinavar F (2004) Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 350(23):2335–2342
Giantonio BJ, Catalano PJ, Meropol NJ, O’Dwyer PJ, Mitchell EP, Alberts SR, Schwartz MA, Benson AB 3rd (2007) Bevacizumab in combination with oxaliplatin, fluorouracil, and leucovorin (FOLFOX4) for previously treated metastatic colorectal cancer: results from the Eastern Cooperative Oncology Group Study E3200. J Clin Oncol 25(12):1539–1544
Sandler A, Gray R, Perry MC, Brahmer J, Schiller JH, Dowlati A, Lilenbaum R, Johnson DH (2006) Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med 355(24):2542–2550
Miller K, Wang M, Gralow J, Dickler M, Cobleigh M, Perez EA, Shenkier T, Cella D, Davidson NE (2007) Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med 357(26):2666–2676
Escudier B, Bellmunt J, Négrier S, Bajetta E, Melichar B, Bracarda S, Ravaud A, Golding S, Jethwa S, Sneller V (2010) Phase III trial of bevacizumab plus interferon alfa-2a in patients with metastatic renal cell carcinoma (AVOREN): final analysis of overall survival. J Clin Oncol 28(13):2144–2150
Tewari KS, Sill MW, Long HJ 3rd, Penson RT, Huang H, Ramondetta LM, Landrum LM, Oaknin A, Reid TJ, Leitao MM, Michael HE, Monk BJ (2014) Improved survival with bevacizumab in advanced cervical cancer. N Engl J Med 370(8):734–743
Pujade-Lauraine E, Hilpert F, Weber B, Reuss A, Poveda A, Kristensen G, Sorio R, Vergote I, Witteveen P, Bamias A, Pereira D, Wimberger P, Oaknin A, Mirza MR, Follana P, Bollag D, Ray-Coquard I (2014) Bevacizumab combined with chemotherapy for platinum-resistant recurrent ovarian cancer: the AURELIA open-label randomized phase III trial. J Clin Oncol 32(13):1302–1308
Lu JF, Bruno R, Eppler S, Novotny W, Lum B, Gaudreault J (2008) Clinical pharmacokinetics of bevacizumab in patients with solid tumors. Cancer Chemother Pharmacol 62(5):779–786
Hoffmann-La Roche LTD F (2014) Investigator’s Brochure Avastin® (bevacizumab). 22nd Version. November 2014
Han K, Jin J, Maia M, Lowe J, Sersch MA, Allison DE (2014) Lower exposure and faster clearance of bevacizumab in gastric cancer and the impact of patient variables: analysis of individual data from AVAGAST phase III trial. AAPS J 16(5):1056–1063
Van Cutsem E, de Haas S, Kang YK, Ohtsu A, Tebbutt NC, Ming Xu J, Peng Yong W, Langer B, Delmar P, Scherer SJ, Shah MA (2012) Bevacizumab in combination with chemotherapy as first-line therapy in advanced gastric cancer: a biomarker evaluation from the AVAGAST randomized phase III trial. J Clin Oncol 30(17):2119–2127
Miles DW, de Haas SL, Dirix LY, Romieu G, Chan A, Pivot X, Tomczak P, Provencher L, Cortés J, Delmar PR, Scherer SJ (2013) Biomarker results from the AVADO phase 3 trial of first-line bevacizumab plus docetaxel for HER2-negative metastatic breast cancer. Br J Cancer 108(5):1052–1060
Beal S, Sheiner LB, Boeckmann A, Bauer RJ (2009) NONMEM user’s guides (1989–2009). Icon Development Solutions, Ellicott City, MD
Lindbom L, Ribbing J, Jonsson EN (2004) Perl-speaks-NONMEM (PsN)—a perl module for NONMEM related programming. Comput Methods Progr Biomed 75(2):85–94
Team RC (2014) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.R-project.org/
Dong JQ, Salinger DH, Endres CJ, Gibbs JP, Hsu CP, Stouch BJ, Hurh E, Gibbs MA (2011) Quantitative prediction of human pharmacokinetics for monoclonal antibodies: retrospective analysis of monkey as a single species for first-in-human prediction. Clin Pharmacokinet 50(2):131–142
Gastonguay MR, French JL, Heitjan DF, Rogers JA, Ahn JE, Ravva P (2010) Missing data in model-based pharmacometric applications: points to consider. J Clin Pharmacol 50(9 Suppl):63S–74S
Karlsson MO, Savic RM (2007) Diagnosing model diagnostics. Clin Pharmacol Ther 82:17–20
Food and Drug Administration (1999) Guidance for industry-population pharmacokinetics. US department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Center for Biologics Evaluation and Research (CBER). http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM072137.pdf
Committee for Medicinal Products for Human Use (CHMP) (2007) Guideline on reporting the results of population pharmacokinetic analyses, Doc. Ref. CHMP/EWP/185990/06. London, 21 June 2007. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003067.pdf
Bergstrand M, Hooker AC, Wallin JE, Karlsson MO (2011) Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J 13(2):143–151
Ette EI (1997) Stability and performance of a population pharmacokinetic model. J Clin Pharmacol 37:486–495
Savic RM (2009) Karlsson MO importance of shrinkage in empirical bayes estimates for diagnostics: problems and solutions. AAPS J 11(3):558–569
Dostalek M, Gardner I, Gurbaxani BM, Rose RH, Chetty M (2013) Pharmacokinetics, pharmacodynamics and physiologically-based pharmacokinetic modelling of monoclonal antibodies. Clin Pharmacokinet 52(2):83–124
Han K, Peyret T, Quartino A, Gosselin NH, Gururangan S, Casanova M, Merks JH, Massimino M, Grill J, Daw NC, Navid F, Jin J, Allison DE (2016) Bevacizumab dosing strategy in paediatric cancer patients based on population pharmacokinetic analysis with external validation. Br J Clin Pharmacol 81(1):148–160
Cobleigh MA et al (2003) A phase I/II dose-escalation trial of bevacizumab in previously treated metastatic breast cancer. Semin Oncol 30(5 Suppl 16):117–124
Kabbinavar F et al (2003) Phase II, randomized trial comparing bevacizumab plus fluorouracil (FU)/leucovorin (LV) with FU/LV alone in patients with metastatic colorectal cancer. J Clin Oncol 21(1):60–65
Hurwitz H et al (2004) Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 350(23):2335–2342
Hillan KJ et al (2003) The role of VEGF expression in response to bevacizumab plus capecitabine in metastatic breast cancer (MBC). In ASCO annual meeting. Abstract No 766
Li J, Gupta M, Jin D, Xin Y, Visich J, Allison DE (2013) Characterization of the long-term pharmacokinetics of bevacizumab following last dose in patients with resected stage II and III carcinoma of the colon. Cancer Chemother Pharmacol 71(3):575–580
Clinical Study Report – BO17704 – A randomized, double-blind multicenter 2-stage phase III study of bevacizumab in combination with cisplatin and gemcitabine versus placebo, cisplatin and gemcitabine in patients with advanced or recurrent non-squamous non-small cell lung cancer, who have not received prior chemotherapy. Roche Report No. 1023798. June 2007
Escudier B et al (2010) Phase III trial of bevacizumab plus interferon alfa-2a in patients with metastatic renal cell carcinoma (AVOREN): final analysis of overall survival. J Clin Oncol 28(13):2144–2150
Escudier B et al (2007) Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma: a randomised, double-blind phase III trial. Lancet 370(9605):2103–2111
Van Cutsem E et al (2009) Phase III trial of bevacizumab in combination with gemcitabine and erlotinib in patients with metastatic pancreatic cancer. J Clin Oncol 27(13):2231–2237
Wu JY et al (2010) Phase I safety and pharmacokinetic study of bevacizumab in Chinese patients with advanced cancer. Chin Med J (Engl) 123(7):901–906
Mok T et al (2014) A correlative biomarker analysis of the combination of bevacizumab and carboplatin-based chemotherapy for advanced nonsquamous non-small-cell lung cancer: results of the phase II randomized ABIGAIL study (BO21015). J Thorac Oncol 9(6):848–855
Chinot O (2012) Phase III trial of bevacizumab added to standard radiotherapy and temozolomide for newly-diagnosed glioblastoma: mature progression-free survival and preliminary overall survival results in AVAglio. In 17th Annual meeting of the society for neuro-oncology. Washington, USA. Abstract No. OT-03
Ito Y et al (2011) Efficacy of first-line bevacizumab (Bev) combined with weekly paclitaxel (wPac) for HER2-negative metastatic breast cancer (MBC): results of a Japanese phase II study (n = 120). In ASCO annual meeting. Abstract No. 1119
Niho S et al (2012) Randomized phase II study of first-line carboplatin-paclitaxel with or without bevacizumab in Japanese patients with advanced non-squamous non-small-cell lung cancer. Lung Cancer 76(3):362–367
Author contributions
All authors have contributed substantially to conception and design of the analysis, drafting or revising the paper as well as giving final approval for submission.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
Kelong Han, Angelica Quartino, Sandhya Girish, David E. Allison and Jin Jin receive salary from Genentech and hold stocks of Roche Pharmaceuticals; in addition, Jin Jin also holds stock in Eli Lilly; Thomas Peyret, Mathilde Marchand and Nathalie H. Gosselin receive salary from Pharsight Consulting Services; all authors declare: no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years; no other relationships or activities that could appear to have influenced the submitted work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Han, K., Peyret, T., Marchand, M. et al. Population pharmacokinetics of bevacizumab in cancer patients with external validation. Cancer Chemother Pharmacol 78, 341–351 (2016). https://doi.org/10.1007/s00280-016-3079-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-016-3079-6