
Abstract
Natural killer (NK) cells are innate lymphoid cells (ILC) known for their ability to recognize and rapidly eliminate infected or transformed cells. Consequently, NK cells are fundamental for host protection against virus infections and malignancies. Even though the critical role of NK cells in cancer immunosurveillance was suspected years ago, the underlying mechanisms took time to be unraveled. Today, it is clear that anti-tumor functions of NK cells are tightly regulated and expand far beyond the simple killing of malignant cells. In spite of tremendous steps made in understanding the NK cell biology, further work is warranted to fully exploit the anticancer potential of these cells. Indeed, tumor-mediated immune suppression hampers NK cell activity, thus complicating their stimulation for therapeutic purposes. Herein, we review the current knowledge of NK cell functions in anti-tumor immunity . We discuss NK cell activity in the cancer immunoediting process with particular emphasis on the elimination and escape phases.
Similar content being viewed by others

Keywords
- Natural Killer
- Natural Killer Cell
- Natural Killer Cell Activity
- Natural Killer Cell Function
- Natural Killer Cell Subset
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Natural killer (NK) cell discovery goes back to the mid-1970s when various groups reported the spontaneous killing of tumor cells by innate lymphoid cells (ILCs) from unimmunized mice (Kiessling et al. 1975; Greenberg and Playfair 1974; Herberman et al. 1975). Interestingly, the idea that these naturally occurring cytotoxic cells might be crucial for cancer immunosurveillance was raised very early (Glimcher et al. 1977; Kiessling et al. 1976). However, at that time, the mechanisms responsible for tumor recognition and elimination remained elusive and it was suggested that NK cells specifically reacted against some antigens of viral origin (Kiessling et al. 1975; Herberman et al. 1975). Ten years later, Ljunggren and Karre conducted a series of experiments leading to the “missing-self” hypothesis which proposes that NK cells recognize and eliminate cells that fail to express self class I molecules of the major histocompatibility complex (MHC-I) (Ljunggren and Karre 1990). Indeed, tumor cells expressing low levels of MHC-I are selectively rejected in vivo in a NK cell-dependent manner (Ljunggren and Karre 1985; Karre et al. 1986). This concept was sufficient to explain the ability of NK cells to eliminate arising malignant cells while sparing normal tissues.
Since then, the comprehension of NK cell regulation and function has dramatically evolved. Nowadays, the name NK refers to a variety of subpopulations with different properties (Hayakawa et al. 2006; Cooper et al. 2001a). In fact, depending on the stimulation, some NK subsets are poor killers but produce massive amount of cytokines and have important regulatory functions (Zhang et al. 2006). In addition, NK cell-mediated killing is far from being spontaneous as it requires prior education (Raulet and Vance 2006) and priming (Long 2007). Therefore, the NK denomination established about 40 years ago is rather misleading and does not reflect the real properties of these cells (Vivier 2006). However, in spite of numerous reconsiderations of NK cell biology, the early speculations on the pivotal role played by NK cells in immunosurveillance proved to be correct.
NK cells’ major role in the control of tumor growth and metastasis has been confirmed by numerous studies performed in rodents such as mice and rats (Smyth et al. 2002). Notably, mice presenting deficiencies in NK cell numbers (Kim et al. 2000; Sathe et al. 2014) or functions (Talmadge et al. 1980) or mice treated with NK cell-depleting antibodies (Seaman et al. 1987) are more susceptible to transplanted tumors and to spontaneous and experimental metastasis. Furthermore, NK cell-depleted mice develop more fibrosarcomas when inoculated with the carcinogen methylcholanthrene (MCA) (Smyth et al. 2001). These observations in animal models are supported by a wide range of clinical data. For instance, patients with high natural cytotoxicity show reduced cancer incidence (Imai et al. 2000). In addition, NK cell infiltration of gastrointestinal sarcoma negatively correlates with the presence of metastases at diagnosis (Delahaye et al. 2011) and high NK cell densities in lung metastases from patients with renal cell carcinoma are associated with improved survival (Remark et al. 2013).
The cancer immunoediting process comprises three phases: elimination, equilibrium, and escape (Dunn et al. 2004). During the elimination phase (i.e., the immunosurveillance phase), effectors from the immune system constantly eradicate new developing tumor cells that have escaped intrinsic tumor suppression. Sometimes the immune system fails to completely eliminate all arising transformed cells, and a dynamic equilibrium is established where new variant tumor clones harbor mutations that render them more resistant to immune attack. Therefore, during the equilibrium phase, the immune system sculpts the tumor population, selecting tumor clones with less immunogenicity. This leads to the escape phase, when the immune system is overwhelmed and very aggressive tumors grow uncontrolled. Unfortunately, most tumors become clinically apparent when already in the escape phase (Vesely et al. 2011). As a result, the phenotype and function of tumor-infiltrating NK cells are already altered at the time of cancer diagnosis (Mamessier et al. 2011; Platonova et al. 2011). An important challenge for the upcoming years is to find ways to reprogram tumor-associated NK cells in order to restore their anti-tumor potential. To this aim, it appears necessary to obtain a full understanding of the mechanisms underlying NK cell anti-tumor activity and the events leading to their alteration. This review will summarize our knowledge about how NK cells protect the host against malignancy and why tumors grow despite such surveillance.
2 Tumor Elimination by NK Cells
2.1 NK Cell Activation
2.1.1 Direct Recognition of Tumor Cells by NK Cells
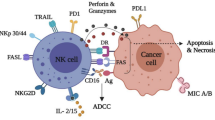
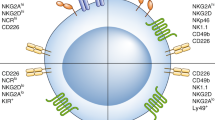
Main activating and inhibitory receptors NK cells express a large array of germline-encoded receptors that allow them to discriminate between normal and altered self (Fig. 1) (Vivier et al. 2008). NKG2D or the natural cytotoxicity receptors (NCR) NKp30 , NKp44 , and NKp46 initiate activating signaling cascades upon binding to stress-induced ligands expressed on the target cell. Conversely, other receptors recognize self-MHC-I- or MHC-I-related molecules at the surface of healthy autologous cells and deliver negative signals. Mouse and human NK cells share the expression of the CD94-NKG2A heterodimer, whereas the human killer cell immunoglobulin-like receptors (KIRs ) and the mouse lectin-like Ly49 receptors have evolved independently in both species (Parham 2005). By blocking activation signals, inhibitory receptors from these different families prevent autoreactivity. Indeed, the balance between positive and negative signals triggered by antagonist receptors determines the initiation of the cytolytic programs. Thus, NK cells recognize and eliminate malignant cells that have lost MHC-I expression (Karre et al. 1986) or upregulated stress-induced molecules (Diefenbach et al. 2001; Cerwenka et al. 2001).

NK cell activation. NK cell activity is controlled by the dynamic integration of activating and inhibitory signals generated by the binding of various surface receptors to ligands expressed by the target cell. Receptor–ligand pairs found in humans only are depicted in orange, the ones in mice only are represented in blue, and molecules expressed by both species are in purple. Note that the list of receptors represented here is not exhaustive and some other receptors have also been involved in target cell recognition by NK cells. In addition to receptor–ligand interactions, cytokines produced by dendritic cells (DCs), monocytes, and macrophages (Mo) or T cells favor NK cell functions. Alternatively, ligands expressed by DCs or monocytes also stimulate NK cells. Finally, pathogen-associated molecular patterns (PAMPs) or danger-associated molecular patterns (DAMPs) potentiate NK cell functions either directly or indirectly by activating DCs
In vivo studies demonstrated a direct involvement of inhibitory and activating NK cell receptors in immunosurveillance . Indeed, the increased development of MCA-induced fibrosarcomas and earlier onset of Eμ-myc-driven lymphomas in mice expressing low levels of Ly49 highlight the importance of missing-self recognition (Tu et al. 2014). Moreover, NKG2D deficiency results in a higher incidence of highly malignant prostate adenocarcinomas and accelerates the progression of Eμ-myc-induced lymphomas (Guerra et al. 2008). Similarly, the absence of NKp46 impairs the eradication of the lymphoma cell line PD1.6 (Halfteck et al. 2009) and increases spontaneous metastasis of B16F10.9 and Lewis lung carcinoma cells (Glasner et al. 2012). However, caution should be taken when interpreting these data because the expression of these activating receptors is not specific of NK cells. In fact, NKG2D expression is also shared by T and NKT cells (Raulet 2003), and albeit more restricted to NK cells, NKp46 is also expressed on other ILCs (Satoh-Takayama et al. 2008) and a discrete T cell subset (Walzer et al. 2007).
Coreceptors The killing of a target cell often requires cooperation between multiple activating signals (Pende et al. 2001; Vitale et al. 1998; Bryceson et al. 2006), or the delivery of an activating signal in the absence of inhibitory signal (Sivori et al. 1999). In addition to the main triggering receptors, some costimulatory receptors or adhesion molecules can potentiate NK cell functions (Biassoni et al. 2001). Such coreceptors are unable to trigger cytotoxicity by their own but dramatically increase NK cell functions when simultaneously engaged with NKG2D or a NCR . This is the case of human 2B4 , where its cross-linking increases target cell lysis upon engagement of NKp46 (Sivori et al. 2000). CD27 and CD28 constitute other examples of costimulatory molecules able to facilitate NK cell-dependent rejection of MHC-I-deficient tumors expressing their respective ligands CD70 and CD80 (Kelly et al. 2002a, b). Finally, DNAX accessory molecule (DNAM-1 or CD226 ) is an adhesion molecule whose ligation synergizes with NCRs for the killing of leukemia cells (Pende et al. 2005). The two CD226 ligands, Nectin-2 (CD112) and the poliovirus receptor (PVR, CD155), are overexpressed in certain human solid and lymphoid malignancies (de Andrade et al. 2014). The importance of CD226 in anti-tumor responses has been demonstrated in vivo (Gilfillan et al. 2008; Tahara-Hanaoka et al. 2006; Croxford et al. 2013). Noteworthy, CD226 -deficient mice are highly susceptible to carcinogen-induced fibrosarcomas or papillomas (Iguchi-Manaka et al. 2008). Interestingly, although CD226 is critical for NK cell-mediated protection against poorly immunogenic tumors, CD226 –CD155 interactions become ineffective when a strong activating receptor such as NKG2D is triggered (Chan et al. 2010; Gilfillan et al. 2008).
In addition to the main inhibitory receptors that bind self-MHC-I molecules, some other surface receptors negatively regulate NK cell functions. One of them, TIGIT , shares its two ligands CD155 and CD112 with the costimulatory molecule CD226 (de Andrade et al. 2014; Martinet and Smyth 2015). In addition, a recent study demonstrated that a third NK cell receptor, CD96 (TACTILE), competes with CD226 for the binding of CD155 and negatively controls cytokine responses by NK cells (Chan et al. 2014). Importantly, CD96−/− mice were found to be more resistant to MCA-induced carcinogenesis and to experimental lung metastasis in a number of tumor models. Thus, interactions of the immunoglobulin-like family members CD226 , TIGIT , and CD96 with CD155 may have significant implications for NK cell-mediated surveillance of tumors.
In spite of being activated in human mature NK cells, 2B4 inhibits the functions of immature human NK cells (Sivori et al. 2002). Intriguingly, mouse 2B4 dampens the activity of mature NK cells, and 2B4 -deficient mice show increased peritoneal clearance of MHC-I-deficient cells expressing 2B4 ligand CD48 (Lee et al. 2004). In addition, compared with CD48− B16 melanoma cells, CD48+ cells are poorly rejected by wild-type mice, suggesting that the expression of CD48 on tumor cells inhibits NK cell-mediated killing (Vaidya et al. 2005). Because CD48 is expressed by all nucleated hematopoietic cells (Kumar and McNerney 2005), a better understanding of 2B4 -mediated regulation of NK cell functions could have important clinical implications for the treatment of blood malignancies.
CD16 and antibody-dependent cell-mediated cytotoxicity NK cells express the low-affinity Fc receptor CD16 (FcγRIIIa) which binds to the constant (Fc) region of immunoglobulins (Caligiuri 2008). CD16 is not a natural cytotoxicity receptor, but it rather enables NK cells to lyse antibody-coated cells, a process called antibody-dependent cell-mediated cytotoxicity (ADCC ) (Santoni et al. 1979). Fc receptors are expressed by the majority of mouse NK cells (Herberman et al. 1977), and 90 % of human blood NK cells are CD16 + (Cooper et al. 2001a). Unlike the other NK cell-activating receptors that require synergistic signals, CD16 cross-linking is sufficient to trigger both cytotoxicity and cytokine release by resting human NK cells (Bryceson et al. 2006). ADCC is possibly a negligible mechanism of cancer immunosurveillance because it relies on the prior elicitation of an adaptive response leading the production to tumor-specific antibodies. However, this mechanism has important implications for the development of tumor- or immune-specific monoclonal antibody-based targeted therapies (Weiner et al. 2010; Pahl et al. 2012; Kohrt et al. 2014).
2.1.2 Contribution of Dendritic Cells and Environmental Factors to NK Cell Activation
NK cell activation by antigen-presenting cells Although NK cells have been named for their spontaneous cytotoxicity toward tumor cells, several pieces of evidence indicate that they require further signals to efficiently eliminate malignant cells (Lucas et al. 2007; Koka et al. 2004). For example, the engagement of a single activating receptor is sufficient to activate IL-2-stimulated NK cells but not resting NK cells (Bryceson et al. 2006). Antigen-presenting cells such as dendritic cells (DCs), monocytes, or macrophages trigger NK cell cytolytic activity and IFN-γ production through cell-to-cell contacts or via the secretion of soluble factors (Degli-Esposti and Smyth 2005; Newman and Riley 2007). Of note, T cells represent an additional source of activating factors (Bihl et al. 2010; Shimizu et al. 2011). Cytokines known to promote NK cell differentiation and activation include IL-2, IL-12, IL-15, IL-18, IL-21, and type I IFN (Degli-Esposti and Smyth 2005). Accordingly, exogenous administration of IL-2, IL-12, IL-18, or IL-21 facilitates NK cell-mediated tumor elimination (Smyth et al. 2000a, 2004; Brady et al. 2004), while mice overexpressing IL-15 show increased protective NK cell activity against MHC class I-negative melanoma cells (Yajima et al. 2002). Conversely, intrinsic type I IFN signaling is not essential for cancer surveillance by mature NK cells (Mizutani et al. 2012), and NK cell depletion does not abrogate some type I IFN-dependent rejection of immunogenic sarcomas (Diamond et al. 2011). Thus, the role of type I IFN in NK cell-mediated anticancer responses, if any, remains to be demonstrated. Finally, cell surface molecules expressed by antigen-presenting cells known to support NK cell activity include CD80, CD86, CD70, CD48 (Degli-Esposti and Smyth 2005), and INAM (Kasamatsu et al. 2014).
In line with the pioneering study by Fernandez and colleagues demonstrating that DCs initiate the regression of MHC-I-deficient tumors in a NK cell-dependent manner (Fernandez et al. 1999), recent data indicate that impaired DC capacity to detect malignant cells and to subsequently activate NK cells results in ineffective tumor control (Chiba et al. 2014). Actually, DC dysfunction in aging mice hinders NK cell-mediated elimination of MHC-I-deficient cells (Guo et al. 2014). Similarly, a deficient cross talk between NK cells and DCs could explain the higher tumor susceptibility of patients suffering from Wiskott–Aldrich syndrome (WAS) (Romagnani and Babic 2014). Therefore, most of the time, NK cells do not respond autonomously to transformed cells, but their ability to protect the host against developing tumors relies on the activity of other immune cells and particularly on DCs.
Toll-like receptors and danger-associated molecular patterns Toll-like receptors (TLRs) are a family of innate sensors that alert immune cells upon recognition of pathogen- or danger-associated molecular patterns (respectively PAMPs and DAMPs) (Janeway and Medzhitov 2002). NK cells express several TLRs, including TLR3, TLR4, and TLR9 that recognize viral nucleic acids or bacterial lipopolysaccharide (LPS) (Adib-Conquy et al. 2014). TLR ligands alone are unable to activate NK cells, but they rather confer some costimulatory signals that synergize with DC or cytokine stimulation (Sivori et al. 2004). Although the relevance of direct DAMP recognition by NK cells is obvious in an infectious context, it is less evident for cancer immunosurveillance because tumors display no or very few danger signals. Nonetheless, such signals can be induced by anticancer treatments. In this regard, tumor cells dying following chemotherapy or radiotherapy release DAMPs that elicit potent immune responses (Apetoh et al. 2007). In addition, TLR agonists are promising adjuvants for cancer immunotherapy, and their ability to efficiently activate NK cells may dramatically influence their therapeutic success (Akazawa et al. 2007; Chin et al. 2010; Zhao et al. 2014).
2.2 Mechanisms of Tumor Elimination by NK Cells
NK cells carry out their anti-tumor function through two main mechanisms: the direct killing of malignant cells and the secretion of IFN-γ (Fig. 2). Alternatively, NK cells modulate the activity of other immune cells. Importantly, two main NK cell subsets have been described in humans: 90 % of blood NK cells are CD56dimCD16hi, whereas the remaining 10 % exhibit high levels of CD56 and low or no expression of CD16 (Cooper et al. 2001a). CD56dim CD16+ cells exhibit greater cytotoxic capacities, while CD56bright cells are a primary source of immunoregulatory cytokines , including IFN-γ, TNF-β, IL-10, IL-13, and GM-CSF (Cooper et al. 2001b; Wilk et al. 2008). Of note, this functional dichotomy has been questioned since the two subsets appear to have equal intrinsic ability to kill or to secrete cytokines but differ in their activation pathways: CD56dim cells are better responders to target cells, and CD56bright cells are more sensitive to cytokines (Vivier 2006). In spite of various attempts, equivalent NK cell subsets have not been identified in mice (Hayakawa et al. 2006; Wilk et al. 2008).

Tumor elimination by NK cells. NK cells directly kill tumor cells by releasing lytic granules containing perforin and granzymes or via the death receptor pathway involving TRAIL or FasL. In addition, NK cells secrete large amount of INF-γ. IFN-γ directly inhibits tumor growth and promotes innate and adaptive immune responses. IFN-γ stimulates the tumoricidal capacities of macrophages (Mo) and favors Th1 polarization of CD4 T cells. Moreover, NK cells eliminate immature DCs or facilitate their maturation, thus allowing the priming of efficient anti-tumor T cell responses by mature DCs
2.2.1 Direct Killing of Tumor Cells by NK Cells
Akin to T lymphocytes, NK cells mediate their cytotoxic activity through the release of lytic granules or via the binding of death receptors.
The granule exocytosis pathway Cytotoxic lymphocytes such as CD8+ T cells and NK cells directionally release granules containing perforin and granzymes upon encounter with a target cell. Perforin creates pores into the membranes of the target cells, thus allowing granzymes and other cytotoxic molecules to access to the cytosol and induce the apoptosis of the target cell (Voskoboinik et al. 2006). The granule exocytosis pathway is considered as the major way of killing by cytotoxic lymphocytes (Kagi et al. 1994; van den Broek et al. 1996). In this regard, NK cells from perforin-deficient mice are unable to lyse YAC-1 or RMA-S target cells in vitro (van den Broek et al. 1995; Kagi et al. 1994). Moreover, in contrast to wild-type mice, perforin-deficient mice fail to reject MHC-I-deficient cells (van den Broek et al. 1995). Perforin plays a critical role in NK cell-mediated protection from tumor metastasis (Smyth et al. 1999), and perforin-dependent cytotoxicity protects against viral and chemical carcinogenesis in vivo (van den Broek et al. 1996). Yet, the greater sensitivity of perforin-deficient mice to transferred, induced, or spontaneous tumors is not necessarily caused by defective NK cell-mediated killing since perforin-mediated CD8+ T cell functions are also involved (Kagi et al. 1994; Smyth et al. 2000b; van den Broek et al. 1996). That said, perforin-deficient mice are more susceptible to MCA-induced fibrosarcomas (Smyth et al. 2000b), a tumor known to be controlled by NK cells (Smyth et al. 2001), and the observation that CD8 deficiency does not confer more susceptibility than the absence of perforin suggests that NK cells are the main effector of perforin-mediated cytotoxicity in this model (van den Broek et al. 1996). If all reports agree on perforin being an essential component of NK cell-mediated immunosurveillance , the role of granzymes is controversial. Some groups demonstrated that granzyme B is necessary for the rapid induction of apoptosis of target cells by NK cells in vitro (Shresta et al. 1995) and that granzymes A and B are required for the in vivo clearance of MHC-I-deficient tumor cells (Pardo et al. 2002). By contrast, another study reported that granzymes A and B do not contribute to in vitro or in vivo NK cell killing, nor are they critical for the control of MCA-induced fibrosarcomas (Davis et al. 2001). The reasons for such discrepancies are unclear. The presence of granzymes could influence the kinetic of killing or the type of cell death (apoptosis versus necrosis), and their requirement might depend on the context. It remains to be established whether the membrane damage induced by perforin is sufficient to cause cell death and whether collective molecules other than granzymes A and B may also be involved.
The Death Receptor pathway Fas ligand (FasL ) and TNF-related apoptosis inducing ligand (TRAIL ) belong to the TNF family (Smyth and Johnstone 2000). These molecules have been shown to mediate NK cell killing of malignant cells (Smyth et al. 2002). While the perforin-mediated pathway can potentially kill any target cell, FasL- and TRAIL-mediated pathways are dependent on the expression of their respective death receptors. Interestingly, NK cells have the capacity to induce Fas expression on malignant cells (Screpanti et al. 2001). Besides, freshly isolated human and mouse NK cells express FasL on their surface and are able to kill Fas-expressing target cells (Oshimi et al. 1996; Arase et al. 1995). In vivo, FasL contributes to the protection against some specific tumor cell lines (Takeda et al. 2001). Yet, a role for FasL usually becomes apparent only when the perforin pathway is defective (van den Broek et al. 1996; Screpanti et al. 2001). Despite its contribution to T cell-mediated surveillance of B cell lymphomas (Afshar-Sterle et al. 2014), several lines of evidence indicate that FasL is rather negligible in NK cell-mediated immunosurveillance (Smyth et al. 1998, 1999). By contrast, TRAIL makes a substantial contribution to NK cell protection against cancer. TRAIL expression has been reported on immature human and mouse NK cells (Zamai et al. 1998; Takeda et al. 2005). TRAIL is also expressed on a population of NK cells in the adult liver that has been recently characterized as a distinct lineage of ILCs (Takeda et al. 2001; Daussy et al. 2014; Seillet et al. 2014). Furthermore, TRAIL expression on NK cells can be induced by different cytokines, including IFN-γ (Takeda et al. 2001), IL-2, and IL-15 (Kayagaki et al. 1999). TRAIL significantly contributes to NK cell-mediated protection against metastasis and prevents the development of carcinogen-induced and spontaneous tumors (Takeda et al. 2001; Cretney et al. 2002; Takeda et al. 2002). Given its preferential expression in neonates, TRAIL-mediated surveillance may be particularly important in early life, before effector molecules of the mature immune system take over (Takeda et al. 2005).
2.2.2 Anti-tumor Activities of IFN-γ
IFN-γ is a pleiotropic cytokine proven to be absolutely critical for cancer immunosurveillance (Ikeda et al. 2002). On the one hand, IFN-γ improves tumor cell immunogenicity, reduces their proliferation, directly induces their apoptosis, and inhibits angiogenesis. On the other hand, IFN-γ activates innate and adaptive immune cells to efficiently fight cancer development. The observation that IFN-γ-deficient mice spontaneously develop lymphomas and lung carcinomas illustrates the pivotal role of IFN-γ in the protection against malignancies (Street et al. 2002). NK cells are major producers of IFN-γ, and its secretion can be stimulated by cytokines such as IL-12 and IL-18 and by the recognition of target cells. In a model of spontaneous metastasis, deficiency in either perforin or IFN-γ increased the tumor burden; but the conjoint deletion of these two components further augmented the number of metastases to similar levels to those obtained following NK cell depletion (Street et al. 2001). Thus, IFN-γ secretion and perforin-mediated cytotoxicity are two independent mechanisms that together fully account for the anti-metastatic activity of NK cells. The interesting observation that TRAIL expression on NK cells is highly dependent on IFN-γ (Takeda et al. 2001, 2002, 2005) suggests that TRAIL could be at least partly responsible for the IFN-γ-dependent pathway of NK cell-mediated anti-tumor immunity. It is important to note that NK cells are not the only components of the innate immune system able to produce IFN-γ. Interestingly, NKT cells, an immune subset that recognizes glycolipids at the surface of transformed cells, have the ability to rapidly activate NK cells through the secretion of IFN-γ (Carnaud et al. 1999; Eberl and MacDonald 2000). The relative importance of NK and NKT cells in promoting IFN-γ and perforin-dependent tumor rejection varies among the models (Street et al. 2001).
It is interesting to note that the most relevant pathway for NK cell-mediated tumor rejection may depend on the way NK cells have been activated. For instance, the elimination of metastases by IL-21-stimulated NK cells is perforin dependent but does not require IFN-γ, TRAIL, or FasL (Brady et al. 2004). Curiously, IL-18 therapy mediates its NK cell activity via FasL or TRAIL and does not depend on the NKG2D-NKG2D ligand, whereas IL-2 and IL-12, which employ perforin-mediated cytotoxicity, appear more effective against metastases expressing NKG2D ligands (Smyth et al. 2004). Therefore, care should be taken in the design of cytokine therapies to ensure that the tumor is sensitive to the targeted pathways.
2.2.3 NK Cells Induce and Orient Adaptive Responses Against Tumors
Tumor-specific T cells are absolutely fundamental for long-term protection. Considered as a first line of defense against cancer, NK cells not only detect malignant cells but also shape adaptive immunity and allow the development of appropriate T cell responses. As mentioned earlier, IFN-γ production by NK cells influences adaptive immunity. For instance, IFN -γ favors the Th1 polarization of CD4+ helper cells (Martin-Fontecha et al. 2004). Other NK cell-derived cytokines and chemokines that significantly modulate immune responses include TNF-α, IL-10, GM-CSF, G-CSF, IL-3, MCP-1 (CCL2), MIP1-α (CCL3), MIP1-β (CCL4), RANTES (CCL5), lymphotactin (XCL1), and IL-8 (CXCL8) (Vivier et al. 2011).
Interestingly, the elimination of transformed cells by NK cells promotes T cell immunity. Indeed, NK cells are not only required for the eradication of MHC-I-deficient tumor cells but also necessary for the development of tumor-specific cytotoxic and Th1 responses that are protective against parental, MHC-I-sufficient cell lines (Kelly et al. 2002a, b). In this context, IFN-γ is essential for the priming of specific T cell responses. Nevertheless, the relevance of these findings for cancer immunosurveillance is unclear since CD8 T cell responses would only have negligible impact on MHC-I-deficient tumors. Yet, NK cell recognition of MHC-Ilow cells and subsequent evoking of T cell immunity could be particularly relevant if only some clones happen to downregulate MHC-I, while the vast majority of the tumor cell population remains MHC-I positive.
NK cells have the ability to “edit” adaptive immune responses by either killing immature DCs or promoting their maturation (Moretta et al. 2005). These steps avoid antigen presentation by immature DCs that could lead to a tolerogenic response. Few in vivo observations support the concept that NK cells eliminate non-immunogenic DCs: Perforin and IFN-γ production are both needed for the expansion of antigen-specific CD8 T cells directed against immunogenic tumors (Strbo et al. 2003), and subcutaneous injection of MHC-I-deficient cells induces a NK cell-dependent reduction of DC numbers in the draining lymph node (Morandi et al. 2012). By contrast, multiple pieces of evidence indicate that NK cells promote DC maturation and antigen presentation. An in vivo study established that the recognition of MHC-Ilow tumor cells stimulates NK cells to prime DCs for IL-12 production and induces highly protective CD8 T cell responses (Mocikat et al. 2003). In addition, IFN-γ enhances the expression of 4-1BB (CD137) on DCs (Pan et al. 2004). In this context, NK cells were found to be essential for CD8 T cell induction and the regression of murine hepatic tumors when IL-12 gene therapy was combined with anti-CD137 costimulation. Recently, LIGHT, a member of the TNF superfamily, was found to be upregulated on the surface of human NK cells following interaction with tumor cells and to participate in NK cell induction of DC maturation (Holmes et al. 2014).
NK-DC cross talk may have important implications for the efficacy of monoclonal antibody-based therapies that target tumor antigens (Lee et al. 2011). Indeed, when tumor cells are coated by rituximab (anti-CD20) or trastuzumab (anti-Her2/neu), NK cells are activated to trigger the presentation of tumor antigens by DCs in an IFN-γ- and TNF-α-dependent manner (Deauvieau et al. 2015). Similarly, cetuximab (anti-EGFR/HER1) bound on the surface of tumor cells activates NK cells through CD16. Stimulated NK cells then facilitate DC maturation and priming of CD8 T cells. As a result, head and neck cancer patients treated with cetuximab have higher frequencies of circulating EGFR-specific CD8 T cells (Srivastava et al. 2013).
Finally, besides direct interaction with DCs, NK cells contribute to adaptive responses in different ways. For instance, NK cell-mediated killing of malignant cells generates cellular debris that constitute antigenic material internalized by antigen-presenting cells. Moreover, by secreting IFN-γ , NK cells may increase the expression of MHC-I by transformed cells and thus facilitate their recognition by cytotoxic CD8 T cells (Shankaran et al. 2001).
2.3 Recruitment of NK Cells into the Tumor Microenvironment
NK cells mainly circulate in the blood where they account for approximately 15 % of lymphocytes in humans (Cooper et al. 2001a). However, NK cells have been observed in several tissues, including the bone marrow, spleen, lymph nodes, liver, lung, omentum, intestine, and placenta (Vivier 2006). Following viral or bacterial assaults, NK cells selectively accumulate at the infection site (Holmberg et al. 1981; McIntyre and Welsh 1986). By contrast, their accumulation is rather variable during malignant transformation. NK cells are enriched in the non-small cell lung carcinoma tumor microenvironment (Platonova et al. 2011) and they represent a substantial percentage (25 %) of the tumor-infiltrating lymphocytes in gastrointestinal sarcoma (Delahaye et al. 2011). By contrast, a large majority of melanoma, hepatocellular carcinoma, breast cancer, and renal cell carcinoma lack significant CD56+ infiltrate (Sconocchia et al. 2012). Thus, the density of NK cell infiltrates within the primary tumor, and metastases vary according to the origin of malignant cells (Remark et al. 2013). Interestingly, in non-small cell lung cancers, NK cells mainly localize in the invasive margin of the cancer, but they are rarely in direct contact with tumor cells and are found outside the tertiary lymphoid structures (Platonova et al. 2011; Carrega et al. 2008).
The mechanisms governing NK cell accumulation at the tumor site are poorly understood. First, it is not clear whether these cells are recruited from the circulation or whether they represent tissue-resident NK cells. Mouse models have allowed the identification of several factors influencing NK cell migration to the site of tumor inoculation. TNF-α mediates NK cell recruitment to the peritoneum in response to intraperitoneal challenge with MHC-I-deficient tumor cells (Smyth et al. 1998). Very surprisingly, the immunosuppressive cytokine IL-10 reportedly favors NK cell accumulation in subcutaneous B16 tumors (Zheng et al. 1996). Ligands expressed on the target cells can also influence NK cell trafficking (Cretney et al. 1999). Indeed, the absence of MHC-I molecules or the presence of NKG2D ligands favors NK cell accumulation (Glas et al. 2000; Diefenbach et al. 2001), whereas experiments using NKp46−/− mice excluded a role for NKp46 ligands (Halfteck et al. 2009). In addition, the same soluble factors that control NK cell migration to an infected/inflamed site are probably involved in cancer. In this aspect, the chemokine receptors CCR2, CCR5, CXCR3, and CX3CR1 regulate NK cell responses to the inflammatory cytokines CCL2, CCL3, CCL5, CCL7, CCL9, CCL11, CCL13, CXCL9, CXCL10, CXCL11, and CX3CL1 (Gregoire et al. 2007). Interestingly, IFN-γ was found to induce local expression of CXCL9, CXCL10, and CXCL11, thus enabling NK cell recruitment in a CXCR3-dependent manner (Wendel et al. 2008). Consequently, numbers of tumor-infiltrating NK cells are severely diminished in mice lacking either CXCR3 or the receptor for IFN-γ. Finally, chemerin is a chemoattractant protein which, in mice, drives the accumulation of NK cells when expressed within the tumor microenvironment (Pachynski et al. 2012). This axis is also likely to be involved in humans because CD56lowCD16+ NK cells express ChemR23, the chemerin receptor (Parolini et al. 2007). In fact, the decreased chemerin expression in several human tumors compared with normal tissues suggests that most malignancies hijack this pathway to escape NK cell-mediated surveillance (Pachynski et al. 2012).
It should be mentioned that the different NK cell subsets possess quite distinct migratory capacity. Indeed, in humans, the CD56bright subset expresses CCR7 and L-selectin (CD62L) and localizes in the lymph nodes, in opposition to the CD56dim subset that represents the vast majority of blood NK cells (Vivier 2006). In addition to CCR7, the CD56bright subset expresses CCR5 and CXCR3, whereas CD56dim NK cells are CXCR1hi and CX3CR1hi (Vitale et al. 2014). As stated earlier, most of these receptors control NK cell recruitment to inflamed tissues. Their differential expression by the CD56bright and CD56low populations implies that one of these two subsets could be preferentially recruited, depending on the tumor microenvironment. In this regard, in non-small lung cell carcinoma, the isolated intratumor NK cells are mainly CD56dim in most patients, but a minor proportion of patients exhibit CD56bright cells (Platonova et al. 2011). In breast cancer, more CD56bright NK cells were found in the tumor compared with the healthy mammary tissue (Mamessier et al. 2011). Similarly, compared with peripheral blood, the proportion of CD56bright NK cells is increased in peritoneal effusions from ovarian cancer patients (Carlsten et al. 2009). Finally, in mouse, CD27hi NK cells, which under steady-state conditions express CXCR3, were found to preferentially accumulate in the tumor tissue (Wendel et al. 2008).
3 Tumor Escape from NK Cells
The key role of NK cells in the protection against cancer is well established. Yet, the fact that malignancies develop in spite of a competent immune system and particularly in spite of the presence of NK cells indicates that, at one point, anti-tumor NK cell activity somehow becomes ineffective. Modifications at the level of the immune system (immunosuppression ) or of the tumor cells (immunoediting ) contribute to tumor escape (Vesely et al. 2011). Tumor-induced immunosuppression refers to the panoply of changes occurring in immune cells that impede them from eliminating the malignant cells. In this aspect, immune responses are actively suppressed by the tumor microenvironment : Tumor-infiltrating lymphocytes and myeloid cells often harbor an altered phenotype and are particularly inefficient at killing transformed cells but rather contribute to the establishment of a chronic inflammation that sustains tumor growth. Immunoediting, for its part, defines the process of immune selection leading to the appearance of tumor clones that are resistant to immune assault. Thus, tumor immunoediting is the consequence of sequential attacks by the immune system that eradicate the most immunogenic cells, but spare the most aggressive ones. The following section will discuss how both immunosuppression and immunoediting hinder NK cell anti-tumor activity (Fig. 3).

Immunoediting and immunosuppression allow tumor escape from NK cells. During the elimination phase, NK cells recognize and eliminate new arising tumor cells that express stress-induced molecules (i.e., ligands for NK cell-activating receptors) and/or low levels of MHC-I (i.e., ligands for NK cell inhibitory receptors). Tumor variants that are less immunogenic due to the loss of expression of ligands for NK cell-activating receptors and/or the upregulation of inhibitory ligands are spared by NK cells and proliferate. During the escape phase, NK cells are no longer able to control the tumor because of the highly suppressive tumor microenvironment that impairs their functions. TGF-β: transforming growth factor-β. PGE2: prostaglandin E2. IDO: indoleamine 2,3-dioxygenase. MDSC: myeloid-derived suppressor cells
3.1 Cancer-Induced Alteration of NK Cell Functions
The numerous ways by which the tumor microenvironment suppresses NK cell function have been reviewed recently (Vitale et al. 2014). In summary, tolerogenic NK cells can be induced by tumor-infiltrating immune cells including regulatory T cells (Tregs) or myeloid-derived suppressor cells (MDSCs), but also by components of the tumor stroma (e.g., fibroblasts) or by the tumor cells themselves. TGF-β is a main inhibitor of NK cell function, and other factors such as prostaglandin E2 (PGE2) or indoleamine 2,3-dioxygenase (IDO) are also involved.
Several groups described a downregulation of activating receptors on tumor-associated NK cells in various human malignancies (Platonova et al. 2011; Mamessier et al. 2011; Carlsten et al. 2009; Krockenberger et al. 2008). Conversely, the inhibitory receptor CD94/NKG2A is increased on NK cell-infiltrating lung carcinomas (Carrega et al. 2008) and breast cancers (Mamessier et al. 2011). Although all reports agree on an altered NK cell repertoire along with an activated phenotype, the precise pattern of modification varies. For instance, NKG2D expression was found to be decreased in breast cancer (Mamessier et al. 2011), but not in ovarian carcinoma (Carlsten et al. 2009). In addition to receptor alteration, tumor-associated NK cells harbor a low perforin content (Carrega et al. 2008) and reduced expression of cytotoxicity-related molecules such as perforin, granzyme B , and TRAIL (Mamessier et al. 2011). Consequently, tumor-infiltrating NK cells are hyporesponsive and display poor cytotoxic activity and cytokine production following in vitro stimulation with target cells (Platonova et al. 2011; Mamessier et al. 2011). It is interesting to note that even if defective NK cell activity is observed in the peripheral blood of breast cancer patients, the deficiency is much more pronounced for tumor-infiltrating NK cells (Mamessier et al. 2011). Remarkably, perturbations of NK cell repertoire correlate with invasive characteristics and poor prognosis in breast cancer (Mamessier et al. 2011). Thus, changes in NK cell receptor expression are certainly key mechanisms by which the tumor escapes innate immunity. The strong immunosuppression within the tumor microenvironment seems to be responsible for these changes, but alterations also occur at a systemic level, in particular at the latter stages of the disease. It is likely that neoplasm formation induces a confined immunosuppressive milieu that initially suppresses NK cell responses locally. However, as the tumor develops, tumor-mediated immune suppression may spread to peripheral NK cells.
The idea that NK cell responses are suppressed by the tumor microenvironment is supported by in vitro experiments wherein peripheral blood NK cells or NK cells from healthy donors were cocultured with tumor cells. Indeed, healthy NK cells display an altered repertoire after incubation with malignant cells (Carlsten et al. 2009; Platonova et al. 2011). Physical contacts between NK cells and tumor cells (Carlsten et al. 2009) as well as soluble factors such as TGF-β and macrophage migration inhibitory factor (MIF) (Krockenberger et al. 2008; Mamessier et al. 2011) are involved in this process. Concerning the downmodulation of NKG2D , a well-known mechanism is the shedding of NKG2D ligands from the surface of the tumor cell: Soluble NKG2D ligands induce the internalization of NKG2D , thereby reducing NKG2D expression on immune cells and contributing to immune suppression (Baragano Raneros et al. 2014). Finally, hypoxia is a prominent feature of the tumor microenvironment that has been shown to disrupt the expression of NCRs and NKG2D (Balsamo et al. 2013). Accordingly, NK cell cytotoxic activity is dramatically reduced at low concentrations of dioxygen (Sarkar et al. 2013). An interesting study indicated that primary tumor hypoxia favors the accumulation of immature NK cells with reduced cytotoxicity within the premetastatic niche, and this altered NK cell function results in tumor escape (Sceneay et al. 2012). Diverse mechanisms are likely to cooperate for the hypoxia-induced dysfunction of NK cells. In this regard, extracellular adenosine, a metabolite produced in the hypoxic tumor microenvironment, has been shown to inhibit NK cell anti-metastatic functions (Young et al. 2014; Mittal et al. 2014; Beavis et al. 2013). Certainly, hypoxia is a factor that must be taken into consideration for the design of new therapies. In this context, the preincubation of NK cells with IL-2 has been shown to overcome the inhibitory effects of hypoxia, at least in vitro (Sarkar et al. 2013). More generally, the use of activating cytokines such as IL-2, IL-15, and IL-21 may help to restore normal levels of NCR expression on cancer patients’ NK cells (Chretien et al. 2014).
Besides the alteration of NK cell activity, a major obstacle to NK cell eradication of established malignancy is their limited access to the tumor bed. In the case of experimental B16F10 metastasis, NLRP3 expression on immune cells was shown to suppress NK cell recruitment to the lung by impeding the accumulation of CCL5- and CXCL9-secreting myeloid cells (Chow et al. 2012). Moreover, as previously discussed, when NK cells are present, they localize preferentially within the stroma and do not make contact with tumor cells (Platonova et al. 2011; Carrega et al. 2008). Very few NK cells are observed in colorectal carcinomas compared with adjacent tissues (Halama et al. 2011). This rather scarce NK cell infiltration contrasts with the high concentrations of adhesion molecules (ICAM-1 and VCAM-1) and high local levels of NK cell-attracting chemokines detected within the malignant tissue. Thus, unknown mechanisms might exclude NK cells from solid tumors.
Defective NK cell activity could have dramatic impact on the success of monoclonal antibody therapies that have been introduced in the clinic to fight against several malignancies (Weiner et al. 2010). Trastuzumab (Herceptin) is a monoclonal antibody specific for human epidermal growth factor receptor 2 (HER-2/neu) that is expressed by metastatic ovarian carcinoma cells. Even if NK cell ADCC capacities were found intact in a hypoxic environment (Balsamo et al. 2013), reduced CD16 expression on tumor-associated NK cells severely impairs ADCC toward trastuzumab-coated fresh ovarian carcinoma cells (Carrega et al. 2008). Therefore, it is possible that many monoclonal antibodies fail to induce ADCC in cancer patients, despite their ability to efficiently trigger ADCC of healthy donor NK cells.
By killing DCs or directly inhibiting T cell proliferation, NK cells have the power to suppress adaptive immune responses (Zhang et al. 2006). Noteworthy, NK cells can limit T cell responses in autoimmune disorders such as multiple sclerosis. Based on these observations, the existence of a regulatory NK cell subset has been proposed. Such “NK-reg cells”, similarly to Tregs, might exert important regulatory effect on anti-tumor immunity. A recent review highlighted the similarities between tumor-infiltrating or tumor-associated NK cells and the decidual NK cells that play an immunosuppressive role during pregnancy (Bruno et al. 2014). Like decidual NK cells, tumor-infiltrating NK cells could establish a tolerogenic microenvironment and facilitate angiogenesis. Hence, despite important pieces of data underlying the anti-tumor role of NK cells, we must consider the possibility that these cells might become protumorigenic. In fact, tumor-derived IL-18 has been shown to induce the expansion of a subset of regulatory Kit+ NK cells that promote tumor outgrowth (Terme et al. 2012). Thus, the conversion of protective NK cells into immunosuppressive NK cells could occur in specific tumor microenvironments and may mark the transition between the equilibrium and the escape phases of the immunoediting process.
3.2 Evidence for Tumor Cell Editing by NK Cells
The first indication of the existence of NK cell-mediated tumor editing came from cultured cell lines. In fact, in vivo-grown tumors are considerably less sensitive to NK cell-mediated killing than their corresponding in vitro lines (Kiessling et al. 1975). For instance, B16 tumors passaged in vivo and cultured in vitro only for a short time are resistant to NK cell-mediated killing , whereas increasing the in vitro culture time to at least 3 weeks results in the acquisition of sensitivity to NK cell-mediated killing (Talmadge et al. 1980). These early experiments suggested that malignant cells growing in an immune-competent environment lose their sensitivity to NK cell assaults.
Subsequently, many data obtained in animal models supported the idea that NK cell activity sculpts tumor cell phenotype. First, lymphomas derived from perforin-deficient mice are systematically rejected when transplanted into wild-type syngeneic mice (Street et al. 2002). Unlike sarcomas derived from wild-type mice, sarcomas derived from perforin-deficient mice express high levels of the NKG2D ligand Rae-1 (Smyth et al. 2005). Moreover, a study using a spontaneous prostate cancer model reported that NKG2D -deficient mice develop large tumors expressing NKG2D ligands, whereas those ligands are absent on comparable tumors arising in wild-type mice, suggesting that NKG2D -dependent immunoselection favors the loss of NKG2D ligands on early-arising tumors (Guerra et al. 2008). In addition to NKG2D , DNAM-1 is another NK cell receptor that shapes tumor immunogenicity. Fibrosarcoma cells that develop in MCA-treated DNAM-1 -deficient mice reportedly express significantly higher levels of the DNAM-1 ligand CD155 than the same tumors arising in wild-type mice (Iguchi-Manaka et al. 2008). Still, perforin-dependent killing and the expression of the activating receptors DNAM-1 and NKG2D are not specific to NK cells. Hence, in the previous examples, immune cells other than NK cells (especially T cells) probably contribute to tumor editing. Noteworthy, the modulation of NKp46 ligand expression on MCA-induced tumors clearly illustrates the tumor editing activity of NK cells. In fact, when they develop in a NKp46-deficient host, MCA-induced fibrosarcomas express higher levels of NKp46 ligands (Elboim et al. 2010). Importantly, the presence of NK cells accounts for reduced tumor growth when these NKp46 -ligand-expressing fibrosarcomas are transplanted into wild-type mice. Furthermore, TRAIL -sensitive MCA-induced sarcomas preferentially develop in NK cell-depleted mice, whereas comparable tumors arising in wild-type mice are generally resistant to TRAIL -mediated killing (Takeda et al. 2002). These data indicate that TRAIL -mediated killing is an important mechanism whereby NK cells eradicate nascent tumor cells during the elimination phase. Finally, the proof that NK cells could manifest cancer immunoediting activity in the absence of adaptive immunity was provided by the direct comparison of carcinogen-induced tumors arising in RAG2−/− mice (i.e., in the absence of adaptive immunity) and in RAG2−/− × γc−/− mice (which lack all lymphocytes including NK cells) (O’Sullivan et al. 2012). Tumors generated in RAG2−/− × γc−/− mice were found to be more immunogenic than those arising in RAG2−/− mice. The immunoediting process occurring in RAG2−/−mice depends on NK cells and IFN-γ production that sustain the accumulation of tumoricidal macrophages. Altogether, studies performed in mice reveal that NK cells do sculpt the emerging neoplasms and reduce their immunogenicity.
Several pieces of evidence indicate that the concept of NK cell-mediated editing of malignant cells described in animal models is also valid in humans. In particular, the loss of ligands for NK cell-activating receptors and/or the upregulation of inhibitory molecules in advanced diseases is consistent with the notion of immunoselection. In colorectal cancer, high levels of NKG2D ligands are detected in early-stage tumors, but their expressions become less and less frequent as the disease progresses (McGilvray et al. 2009). In addition, HLA-G, the ligand for the inhibitory NK receptor KIR 2DL4, is absent on the initial tumor lesions but upregulated in the late stages of different cancers (Urosevic and Dummer 2008). Thus, HLA-G may have a role in the final phase of immunoediting. Importantly, in addition to the local effects of membrane-bound HLA-G, secreted soluble HLA-G molecules may enter the circulation and induce immunosuppression. Lastly, the cancer immunoediting process can also favor the growth of tumor cells that are resistant to NK-mediated killing pathways. In this regard, some human tumor cell lines were found to be insensitive to IFN-γ (Kaplan et al. 1998).
In summary, although T cells define the antigenicity of the tumors, NK cells are not negligible in immunoediting process. Noteworthy, in an immunocompetent host, tumor cells can become apparent if they are able to escape NK cell-mediated immunosurveillance . Consequently, tumor cells that are poorly recognized by NK cells would be preferentially selected. However, the modulation of cell surface molecules is not the only mechanism possible. It is important to underline that some tumors grow despite their high expression of NK cell-activating ligands. Such tumors must have developed other ways to hijack NK cell-mediated control. They might have become resistant to NK cell-mediated killing or induced strong immunosuppression .
4 Concluding Remarks
4.1 Are NK Cells Essential for Tumor Control?
Even if some evidence argues for a role of NK cells in immunosurveillance, the general importance of NK cells in host protection against tumors may not be clear. Numerous studies are biased because they use tumor cell lines previously selected for their sensitivity to NK cells (e.g., YAC-1, RMA-S, B16F10, or Rae1-β-expressing cell lines) to assess NK cell functions in vivo. Although NK cells were shown to control MCA-induced fibrosarcoma (Smyth et al. 2001), they appeared to be negligible for the protection against virally induced fibrosarcoma (van den Broek et al. 1996) or against spontaneously arising lymphoma in perforin-deficient mice lacking one allele of the tumor suppressor p53 (Smyth et al. 2000b). Besides, the relative contribution of NK cells compared with other immune effector cells is not clearly defined, and the increased tumor occurrence in mice lacking IFN-γ , perforin , or some NK cell receptors (e.g., NKG2D or DNAM-1 ) could be the result of defective NKT or T cell functions. Ideally, NK cell-specific deletions of these effector molecules are required or at least for now, transfer of sorted NK cells defective in these pathways into lymphocyte-deficient mice. A mouse model that specifically and constantly lacks NK cells has recently been described: the Mcl1fl/flNcr1-Cre+ mice (Sathe et al. 2014). These mice will help to definitely determine whether NK cells do prevent the spontaneous occurrence of cancer. Of note, NK cells have been recently included into the large family of ILCs (Spits et al. 2013), and some anti-tumor functions previously attributed to NK cells could be performed by distinct ILC subsets. As a matter of fact, the origins of TRAIL+NK1.1+CD3− cells, responsible for the protection against liver metastasis (Takeda et al. 2001), remains a matter of debate (Daussy et al. 2014; Sojka et al. 2014).
Few reported cases of NK cell deficiency provide valuable information on NK cell functions in human (Orange 2013). Patients suffering from NK cell deficiency are definitely more susceptible to herpesvirus or papillomavirus infections. Interestingly, in a cohort of 19 patients, a significant percentage (21 %) experienced malignancies, including Epstein–Barr virus (EBV)- or human papillomavirus (HPV)-driven malignancies and leukemia. Still, this could be attributed to a defective control of virus infections rather than tumor cells. The paucity of cases of NK cell deficiency, the time required for tumors to develop, and the premature death of these patients complicate the interpretation of these data. Therefore, whether NK cell deficiency leads to increased cancer rate or metastasis remains an opened question.
4.2 When and Where Are NK Cells Important?
Since they do not require selection or expansion of specific clones, NK cells together with other innate cells such as γδ T cells or NKT cells represent a first line of defense against malignancy. Nonetheless, the appealing idea that NK cells act as sentinels screening the appearance of transformed cells within the entire body remains to be clearly demonstrated. Different populations of tissue-resident NK cells may perform the sentinel function in specific organs, while circulating NK cells appear to be highly efficient against bloodborne metastases. NK cells are required at the very earliest times of tumor development (Halfteck et al. 2009) and are absolutely crucial for protection from metastases (Sathe et al. 2014). Consistent with their role of first guardians, NK cells protect mice injected with small numbers of tumor cells but are ineffective against large inoculums (Ljunggren and Karre 1990). These data explain why NK cells appear more effective against metastases than primary tumors (Glasner et al. 2012). Still, in some cancers, the presence of NK cells does not impact patient outcome (Platonova et al. 2011). This does not mean that NK cells never had any role in these malignancies but rather indicates that their functions have been turned off. It is tempting to simply speculate that NK cells act at the very beginning of tumor transformation and then T cells become more critical. However, NK cells and T cells have complementary detection mechanisms, and even at later time points, NK cells may be critical in eliminating MHC-Ilow variants that escape T cell surveillance. To go back to the three phases of cancer immunoediting , it is clear that NK cells are fundamental for the elimination phase. However, they do not appear to do much during the equilibrium phase where the tumor is held in check by the adaptive immune system (Koebel et al. 2007). Then, during the escape phase, tumor-altered NK cells either may be mere bystanders or may in the worst case promote tumor progression (Bruno et al. 2014).
4.3 Therapeutic Use of NK Cells
Developing therapies able to restore NK cell anti-tumor activity has been a challenge for many years. This is not an easy task considering that NK cells are really effective only at early time points of cellular transformation and against low tumor burden. Still, the transfer of MHC-I-mismatched NK cells has provided promising results in hematological malignancies (Chan et al. 2008). Regarding solid tumors, NK cells might help restraining residual tumor cells after surgical removal of the primary tumor mass. Promoting NK cell-mediated ADCC would probably benefit patients receiving monoclonal antibody therapies. Treatment with cytokines such as IL-2 and IL-15 can boost NK cell functions and should be preferentially used in combination with chemotherapeutics known to trigger the upregulation of ligands for activating receptors (Soriani et al. 2009). Alternatively, the blockade of inhibitory receptors by anti-KIR monoclonal antibodies may improve NK cytotoxic activity (Benson et al. 2012). Moreover, the adoptive transfer of allogeneic NK cells has shown some success not only against leukemia but also against solid tumors (Eguizabal et al. 2014), and the engineering of NK cells expressing a chimeric antigen receptor (CAR) receives attention (Klingemann 2014). Finally, in the last decade, immune checkpoint blockade has emerged as a powerful way to restore anti-tumor immunity (Pardoll 2012). Although the success of immune checkpoint antibodies has been mainly attributed to their T cell-stimulating capacity, NK cells are likely to be involved. To conclude, administration of therapeutic agents able to restore NK cell functions following the removal of the primary tumor will undoubtedly benefit patients since NK cells would not only control metastatic spread but also promote adaptive responses which are crucial for the establishment of long-lasting protective immunity.
References
Adib-Conquy M, Scott-Algara D, Cavaillon JM, Souza-Fonseca-Guimaraes F (2014) TLR-mediated activation of NK cells and their role in bacterial/viral immune responses in mammals. Immunol Cell Biol 92(3):256–262. doi:10.1038/icb.2013.99
Afshar-Sterle S, Zotos D, Bernard NJ, Scherger AK, Rodling L, Alsop AE, Walker J, Masson F, Belz GT, Corcoran LM, O’Reilly LA, Strasser A, Smyth MJ, Johnstone R, Tarlinton DM, Nutt SL, Kallies A (2014) Fas ligand-mediated immune surveillance by T cells is essential for the control of spontaneous B cell lymphomas. Nat Med 20(3):283–290. doi:10.1038/nm.3442
Akazawa T, Ebihara T, Okuno M, Okuda Y, Shingai M, Tsujimura K, Takahashi T, Ikawa M, Okabe M, Inoue N, Okamoto-Tanaka M, Ishizaki H, Miyoshi J, Matsumoto M, Seya T (2007) Antitumor NK activation induced by the toll-like receptor 3-TICAM-1 (TRIF) pathway in myeloid dendritic cells. Proc Natl Acad Sci USA 104(1):252–257. doi:10.1073/pnas.0605978104
Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, Maiuri MC, Ullrich E, Saulnier P, Yang H, Amigorena S, Ryffel B, Barrat FJ, Saftig P, Levi F, Lidereau R, Nogues C, Mira JP, Chompret A, Joulin V, Clavel-Chapelon F, Bourhis J, Andre F, Delaloge S, Tursz T, Kroemer G, Zitvogel L (2007) Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med 13(9):1050–1059. doi:10.1038/nm1622
Arase H, Arase N, Saito T (1995) Fas-mediated cytotoxicity by freshly isolated natural killer cells. J Exp Med 181(3):1235–1238
Balsamo M, Manzini C, Pietra G, Raggi F, Blengio F, Mingari MC, Varesio L, Moretta L, Bosco MC, Vitale M (2013) Hypoxia downregulates the expression of activating receptors involved in NK-cell-mediated target cell killing without affecting ADCC. Eur J Immunol 43(10):2756–2764. doi:10.1002/eji.201343448
Baragano Raneros A, Suarez-Alvarez B, Lopez-Larrea C (2014) Secretory pathways generating immunosuppressive NKG2D ligands: new targets for therapeutic intervention. Oncoimmunology 3:e28497. doi:10.4161/onci.28497
Beavis PA, Divisekera U, Paget C, Chow MT, John LB, Devaud C, Dwyer K, Stagg J, Smyth MJ, Darcy PK (2013) Blockade of A2A receptors potently suppresses the metastasis of CD73+ tumors. Proc Natl Acad Sci USA 110(36):14711–14716. doi:10.1073/pnas.1308209110
Benson DM Jr, Hofmeister CC, Padmanabhan S, Suvannasankha A, Jagannath S, Abonour R, Bakan C, Andre P, Efebera Y, Tiollier J, Caligiuri MA, Farag SS (2012) A phase 1 trial of the anti-KIR antibody IPH2101 in patients with relapsed/refractory multiple myeloma. Blood 120(22):4324–4333. doi:10.1182/blood-2012-06-438028
Biassoni R, Cantoni C, Pende D, Sivori S, Parolini S, Vitale M, Bottino C, Moretta A (2001) Human natural killer cell receptors and co-receptors. Immunol Rev 181:203–214
Bihl F, Pecheur J, Breart B, Poupon G, Cazareth J, Julia V, Glaichenhaus N, Braud VM (2010) Primed antigen-specific CD4+ T cells are required for NK cell activation in vivo upon Leishmania major infection. J Immunol 185(4):2174–2181. doi:10.4049/jimmunol.1001486
Brady J, Hayakawa Y, Smyth MJ, Nutt SL (2004) IL-21 induces the functional maturation of murine NK cells. J Immunol 172(4):2048–2058
Bruno A, Ferlazzo G, Albini A, Noonan DM (2014) A think tank of TINK/TANKs: tumor-infiltrating/tumor-associated natural killer cells in tumor progression and angiogenesis. J Nat Cancer Inst 106(8): dju200. doi:10.1093/jnci/dju200
Bryceson YT, March ME, Ljunggren HG, Long EO (2006) Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood 107(1):159–166. doi:10.1182/blood-2005-04-1351
Caligiuri MA (2008) Human natural killer cells. Blood 112(3):461–469. doi:10.1182/blood-2007-09-077438
Carlsten M, Norell H, Bryceson YT, Poschke I, Schedvins K, Ljunggren HG, Kiessling R, Malmberg KJ (2009) Primary human tumor cells expressing CD155 impair tumor targeting by down-regulating DNAM-1 on NK cells. J Immunol 183(8):4921–4930. doi:10.4049/jimmunol.0901226
Carnaud C, Lee D, Donnars O, Park SH, Beavis A, Koezuka Y, Bendelac A (1999) Cutting edge: cross-talk between cells of the innate immune system: NKT cells rapidly activate NK cells. J Immunol 163(9):4647–4650
Carrega P, Morandi B, Costa R, Frumento G, Forte G, Altavilla G, Ratto GB, Mingari MC, Moretta L, Ferlazzo G (2008) Natural killer cells infiltrating human nonsmall-cell lung cancer are enriched in CD56 bright CD16(-) cells and display an impaired capability to kill tumor cells. Cancer 112(4):863–875. doi:10.1002/cncr.23239
Cerwenka A, Baron JL, Lanier LL (2001) Ectopic expression of retinoic acid early inducible-1 gene (RAE-1) permits natural killer cell-mediated rejection of a MHC class I-bearing tumor in vivo. Proc Natl Acad Sci USA 98(20):11521–11526. doi:10.1073/pnas.201238598
Chan CJ, Andrews DM, Smyth MJ (2008) Can NK cells be a therapeutic target in human cancer? Eur J Immunol 38(11):2964–2968. doi:10.1002/eji.200838764
Chan CJ, Andrews DM, McLaughlin NM, Yagita H, Gilfillan S, Colonna M, Smyth MJ (2010) DNAM-1/CD155 interactions promote cytokine and NK cell-mediated suppression of poorly immunogenic melanoma metastases. J Immunol 184(2):902–911. doi:10.4049/jimmunol.0903225
Chan CJ, Martinet L, Gilfillan S, Souza-Fonseca-Guimaraes F, Chow MT, Town L, Ritchie DS, Colonna M, Andrews DM, Smyth MJ (2014) The receptors CD96 and CD226 oppose each other in the regulation of natural killer cell functions. Nat Immunol 15(5):431–438. doi:10.1038/ni.2850
Chiba S, Ikushima H, Ueki H, Yanai H, Kimura Y, Hangai S, Nishio J, Negishi H, Tamura T, Saijo S, Iwakura Y, Taniguchi T (2014) Recognition of tumor cells by Dectin-1 orchestrates innate immune cells for anti-tumor responses eLife 3:e04177. doi:10.7554/eLife.04177
Chin AI, Miyahira AK, Covarrubias A, Teague J, Guo B, Dempsey PW, Cheng G (2010) Toll-like receptor 3-mediated suppression of TRAMP prostate cancer shows the critical role of type I interferons in tumor immune surveillance. Cancer Res 70(7):2595–2603. doi:10.1158/0008-5472.CAN-09-1162
Chow MT, Sceneay J, Paget C, Wong CS, Duret H, Tschopp J, Moller A, Smyth MJ (2012) NLRP3 suppresses NK cell-mediated responses to carcinogen-induced tumors and metastases. Cancer Res 72(22):5721–5732. doi:10.1158/0008-5472.CAN-12-0509
Chretien AS, Le Roy A, Vey N, Prebet T, Blaise D, Fauriat C, Olive D (2014) Cancer-induced alterations of NK-mediated target recognition: current and investigational pharmacological strategies aiming at restoring NK-mediated anti-tumor activity. Front Immunol 5:122. doi:10.3389/fimmu.2014.00122
Cooper MA, Fehniger TA, Caligiuri MA (2001a) The biology of human natural killer-cell subsets. Trends Immunol 22(11):633–640
Cooper MA, Fehniger TA, Turner SC, Chen KS, Ghaheri BA, Ghayur T, Carson WE, Caligiuri MA (2001b) Human natural killer cells: a unique innate immunoregulatory role for the CD56 (bright) subset. Blood 97(10):3146–3151
Cretney E, Degli-Esposti MA, Densley EH, Farrell HE, Davis-Poynter NJ, Smyth MJ (1999) m144, a murine cytomegalovirus (MCMV)-encoded major histocompatibility complex class I homologue, confers tumor resistance to natural killer cell-mediated rejection. J Exp Med 190(3):435–444
Cretney E, Takeda K, Yagita H, Glaccum M, Peschon JJ, Smyth MJ (2002) Increased susceptibility to tumor initiation and metastasis in TNF-related apoptosis-inducing ligand-deficient mice. J Immunol 168(3):1356–1361
Croxford JL, Tang ML, Pan MF, Huang CW, Kamran N, Phua CM, Chng WJ, Ng SB, Raulet DH, Gasser S (2013) ATM-dependent spontaneous regression of early Emu-myc-induced murine B-cell leukemia depends on natural killer and T cells. Blood 121(13):2512–2521. doi:10.1182/blood-2012-08-449025
Daussy C, Faure F, Mayol K, Viel S, Gasteiger G, Charrier E, Bienvenu J, Henry T, Debien E, Hasan UA, Marvel J, Yoh K, Takahashi S, Prinz I, de Bernard S, Buffat L, Walzer T (2014) T-bet and Eomes instruct the development of two distinct natural killer cell lineages in the liver and in the bone marrow. J Exp Med 211(3):563–577. doi:10.1084/jem.20131560
Davis JE, Smyth MJ, Trapani JA (2001) Granzyme A and B-deficient killer lymphocytes are defective in eliciting DNA fragmentation but retain potent in vivo anti-tumor capacity. Eur J Immunol 31(1):39–47. doi:10.1002/1521-4141(200101)31:1<39:AID-IMMU39>3.0.CO;2-1
de Andrade LF, Smyth MJ, Martinet L (2014) DNAM-1 control of natural killer cells functions through nectin and nectin-like proteins. Immunol Cell Biol 92(3):237–244. doi:10.1038/icb.2013.95
Deauvieau F, Ollion V, Doffin AC, Achard C, Fonteneau JF, Verronese E, Durand I, Ghittoni R, Marvel J, Dezutter-Dambuyant C, Walzer T, Vie H, Perrot I, Goutagny N, Caux C, Valladeau-Guilemond J (2015) Human natural killer cells promote cross-presentation of tumor cell-derived antigens by dendritic cells. Int J Cancer 136(5):1085–1094. doi:10.1002/ijc.29087
Degli-Esposti MA, Smyth MJ (2005) Close encounters of different kinds: dendritic cells and NK cells take centre stage. Nat Rev Immunol 5(2):112–124. doi:10.1038/nri1549
Delahaye NF, Rusakiewicz S, Martins I, Menard C, Roux S, Lyonnet L, Paul P, Sarabi M, Chaput N, Semeraro M, Minard-Colin V, Poirier-Colame V, Chaba K, Flament C, Baud V, Authier H, Kerdine-Romer S, Pallardy M, Cremer I, Peaudecerf L, Rocha B, Valteau-Couanet D, Gutierrez JC, Nunes JA, Commo F, Bonvalot S, Ibrahim N, Terrier P, Opolon P, Bottino C, Moretta A, Tavernier J, Rihet P, Coindre JM, Blay JY, Isambert N, Emile JF, Vivier E, Lecesne A, Kroemer G, Zitvogel L (2011) Alternatively spliced NKp30 isoforms affect the prognosis of gastrointestinal stromal tumors. Nat Med 17(6):700–707. doi:10.1038/nm.2366
Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, Lee H, Arthur CD, White JM, Kalinke U, Murphy KM, Schreiber RD (2011) Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med 208(10):1989–2003. doi:10.1084/jem.20101158
Diefenbach A, Jensen ER, Jamieson AM, Raulet DH (2001) Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature 413(6852):165–171. doi:10.1038/35093109
Dunn GP, Old LJ, Schreiber RD (2004) The three Es of cancer immunoediting. Annu Rev Immunol 22:329–360. doi:10.1146/annurev.immunol.22.012703.104803
Eberl G, MacDonald HR (2000) Selective induction of NK cell proliferation and cytotoxicity by activated NKT cells. Eur J Immunol 30(4):985–992. doi:10.1002/(SICI)1521-4141(200004)30:4<985:AID-IMMU985>3.0.CO;2-E
Eguizabal C, Zenarruzabeitia O, Monge J, Santos S, Vesga MA, Maruri N, Arrieta A, Rinon M, Tamayo-Orbegozo E, Amo L, Larrucea S, Borrego F (2014) Natural killer cells for cancer immunotherapy: pluripotent stem cells-derived NK cells as an immunotherapeutic perspective. Front Immunol 5:439. doi:10.3389/fimmu.2014.00439
Elboim M, Gazit R, Gur C, Ghadially H, Betser-Cohen G, Mandelboim O (2010) Tumor immunoediting by NKp46. J Immunol 184(10):5637–5644. doi:10.4049/jimmunol.0901644
Fernandez NC, Lozier A, Flament C, Ricciardi-Castagnoli P, Bellet D, Suter M, Perricaudet M, Tursz T, Maraskovsky E, Zitvogel L (1999) Dendritic cells directly trigger NK cell functions: cross-talk relevant in innate anti-tumor immune responses in vivo. Nat Med 5(4):405–411. doi:10.1038/7403
Gilfillan S, Chan CJ, Cella M, Haynes NM, Rapaport AS, Boles KS, Andrews DM, Smyth MJ, Colonna M (2008) DNAM-1 promotes activation of cytotoxic lymphocytes by nonprofessional antigen-presenting cells and tumors. J Exp Med 205(13):2965–2973. doi:10.1084/jem.20081752
Glas R, Franksson L, Une C, Eloranta ML, Ohlen C, Orn A, Karre K (2000) Recruitment and activation of natural killer (NK) cells in vivo determined by the target cell phenotype. An adaptive component of NK cell-mediated responses. J Exp Med 191(1):129–138
Glasner A, Ghadially H, Gur C, Stanietsky N, Tsukerman P, Enk J, Mandelboim O (2012) Recognition and prevention of tumor metastasis by the NK receptor NKp46/NCR1. J Immunol 188(6):2509–2515. doi:10.4049/jimmunol.1102461
Glimcher L, Shen FW, Cantor H (1977) Identification of a cell-surface antigen selectively expressed on the natural killer cell. J Exp Med 145(1):1–9
Greenberg AH, Playfair JH (1974) Spontaneously arising cytotoxicity to the P-815-Y mastocytoma in NZB mice. Clin Exp Immunol 16(1):99–109
Gregoire C, Chasson L, Luci C, Tomasello E, Geissmann F, Vivier E, Walzer T (2007) The trafficking of natural killer cells. Immunol Rev 220:169–182. doi:10.1111/j.1600-065X.2007.00563.x
Guerra N, Tan YX, Joncker NT, Choy A, Gallardo F, Xiong N, Knoblaugh S, Cado D, Greenberg NM, Raulet DH (2008) NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity 28(4):571–580. doi:10.1016/j.immuni.2008.02.016
Guo Z, Tilburgs T, Wong B, Strominger JL (2014) Dysfunction of dendritic cells in aged C57BL/6 mice leads to failure of natural killer cell activation and of tumor eradication. Proc Natl Acad Sci USA 111(39):14199–14204. doi:10.1073/pnas.1414780111
Halama N, Braun M, Kahlert C, Spille A, Quack C, Rahbari N, Koch M, Weitz J, Kloor M, Zoernig I, Schirmacher P, Brand K, Grabe N, Falk CS (2011) Natural killer cells are scarce in colorectal carcinoma tissue despite high levels of chemokines and cytokines. Clinical Cancer Res An Official J Am Assoc Cancer Res 17(4):678–689. doi:10.1158/1078-0432.CCR-10-2173
Halfteck GG, Elboim M, Gur C, Achdout H, Ghadially H, Mandelboim O (2009) Enhanced in vivo growth of lymphoma tumors in the absence of the NK-activating receptor NKp46/NCR1. J Immunol 182(4):2221–2230. doi:10.4049/jimmunol.0801878
Hayakawa Y, Huntington ND, Nutt SL, Smyth MJ (2006) Functional subsets of mouse natural killer cells. Immunol Rev 214:47–55. doi:10.1111/j.1600-065X.2006.00454.x
Herberman RB, Nunn ME, Lavrin DH (1975) Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic acid allogeneic tumors. I. Distribution of reactivity and specificity. Int J Cancer 16 (2):216–229
Herberman RB, Bartram S, Haskill JS, Nunn M, Holden HT, West WH (1977) Fc receptors on mouse effector cells mediating natural cytotoxicity against tumor cells. J Immunol 119(1):322–326
Holmberg LA, Springer TA, Ault KA (1981) Natural killer activity in the peritoneal exudates of mice infected with Listeria monocytogenes: characterization of the natural killer cells by using a monoclonal rat anti-murine macrophage antibody (M1/70). J Immunol 127(5):1792–1799
Holmes TD, Wilson EB, Black EV, Benest AV, Vaz C, Tan B, Tanavde VM, Cook GP (2014) Licensed human natural killer cells aid dendritic cell maturation via TNFSF14/LIGHT. Proc Natl Acad Sci USA 111(52):E5688–E5696. doi:10.1073/pnas.1411072112
Iguchi-Manaka A, Kai H, Yamashita Y, Shibata K, Tahara-Hanaoka S, Honda S, Yasui T, Kikutani H, Shibuya K, Shibuya A (2008) Accelerated tumor growth in mice deficient in DNAM-1 receptor. J Exp Med 205(13):2959–2964. doi:10.1084/jem.20081611
Ikeda H, Old LJ, Schreiber RD (2002) The roles of IFN gamma in protection against tumor development and cancer immunoediting. Cytokine Growth Factor Rev 13(2):95–109
Imai K, Matsuyama S, Miyake S, Suga K, Nakachi K (2000) Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: an 11-year follow-up study of a general population. Lancet 356(9244):1795–1799. doi:10.1016/S0140-6736(00)03231-1
Janeway CA Jr, Medzhitov R (2002) Innate immune recognition. Annu Rev Immunol 20:197–216. doi:10.1146/annurev.immunol.20.083001.084359
Kagi D, Ledermann B, Burki K, Seiler P, Odermatt B, Olsen KJ, Podack ER, Zinkernagel RM, Hengartner H (1994) Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature 369(6475):31–37. doi:10.1038/369031a0
Kaplan DH, Shankaran V, Dighe AS, Stockert E, Aguet M, Old LJ, Schreiber RD (1998) Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc Natl Acad Sci USA 95(13):7556–7561
Karre K, Ljunggren HG, Piontek G, Kiessling R (1986) Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature 319(6055):675–678. doi:10.1038/319675a0
Kasamatsu J, Azuma M, Oshiumi H, Morioka Y, Okabe M, Ebihara T, Matsumoto M, Seya T (2014) INAM plays a critical role in IFN-gamma production by NK cells interacting with polyinosinic-polycytidylic acid-stimulated accessory cells. J Immunol 193(10):5199–5207. doi:10.4049/jimmunol.1400924
Kayagaki N, Yamaguchi N, Nakayama M, Takeda K, Akiba H, Tsutsui H, Okamura H, Nakanishi K, Okumura K, Yagita H (1999) Expression and function of TNF-related apoptosis-inducing ligand on murine activated NK cells. J Immunol 163(4):1906–1913
Kelly JM, Darcy PK, Markby JL, Godfrey DI, Takeda K, Yagita H, Smyth MJ (2002a) Induction of tumor-specific T cell memory by NK cell-mediated tumor rejection. Nat Immunol 3(1):83–90. doi:10.1038/ni746
Kelly JM, Takeda K, Darcy PK, Yagita H, Smyth MJ (2002b) A role for IFN-gamma in primary and secondary immunity generated by NK cell-sensitive tumor-expressing CD80 in vivo. J Immunol 168(9):4472–4479
Kiessling R, Klein E, Wigzell H (1975) “Natural” killer cells in the mouse. I. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Specificity and distribution according to genotype. Eur J Immunol 5(2):112–117. doi:10.1002/eji.1830050208
Kiessling R, Petranyi G, Klein G, Wigzell H (1976) Non-T-cell resistance against a mouse Moloney lymphoma. Int J Cancer 17(2):275–281
Kim S, Iizuka K, Aguila HL, Weissman IL, Yokoyama WM (2000) In vivo natural killer cell activities revealed by natural killer cell-deficient mice. Proc Natl Acad Sci USA 97(6):2731–2736. doi:10.1073/pnas.050588297
Klingemann H (2014) Are natural killer cells superior CAR drivers? Oncoimmunology 3:e28147. doi:10.4161/onci.28147
Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, Smyth MJ, Schreiber RD (2007) Adaptive immunity maintains occult cancer in an equilibrium state. Nature 450(7171):903–907. doi:10.1038/nature06309
Kohrt HE, Colevas AD, Houot R, Weiskopf K, Goldstein MJ, Lund P, Mueller A, Sagiv-Barfi I, Marabelle A, Lira R, Troutner E, Richards L, Rajapaska A, Hebb J, Chester C, Waller E, Ostashko A, Weng WK, Chen L, Czerwinski D, Fu YX, Sunwoo J, Levy R (2014) Targeting CD137 enhances the efficacy of cetuximab. J Clin Investig 124(6):2668–2682. doi:10.1172/JCI73014
Koka R, Burkett P, Chien M, Chai S, Boone DL, Ma A (2004) Cutting edge: murine dendritic cells require IL-15R alpha to prime NK cells. J Immunol 173(6):3594–3598
Krockenberger M, Dombrowski Y, Weidler C, Ossadnik M, Honig A, Hausler S, Voigt H, Becker JC, Leng L, Steinle A, Weller M, Bucala R, Dietl J, Wischhusen J (2008) Macrophage migration inhibitory factor contributes to the immune escape of ovarian cancer by down-regulating NKG2D. J Immunol 180(11):7338–7348
Kumar V, McNerney ME (2005) A new self: MHC-class-I-independent natural-killer-cell self-tolerance. Nat Rev Immunol 5(5):363–374. doi:10.1038/nri1603
Lee KM, McNerney ME, Stepp SE, Mathew PA, Schatzle JD, Bennett M, Kumar V (2004) 2B4 acts as a non-major histocompatibility complex binding inhibitory receptor on mouse natural killer cells. J Exp Med 199(9):1245–1254. doi:10.1084/jem.20031989
Lee SC, Srivastava RM, Lopez-Albaitero A, Ferrone S, Ferris RL (2011) Natural killer (NK): dendritic cell (DC) cross talk induced by therapeutic monoclonal antibody triggers tumor antigen-specific T cell immunity. Immunol Res 50(2–3):248–254. doi:10.1007/s12026-011-8231-0
Ljunggren HG, Karre K (1985) Host resistance directed selectively against H-2-deficient lymphoma variants. Analysis of the mechanism. J Exp Med 162(6):1745–1759
Ljunggren HG, Karre K (1990) In search of the ‘missing self’: MHC molecules and NK cell recognition. Immunol Today 11(7):237–244
Long EO (2007) Ready for prime time: NK cell priming by dendritic cells. Immunity 26(4):385–387. doi:10.1016/j.immuni.2007.04.001
Lucas M, Schachterle W, Oberle K, Aichele P, Diefenbach A (2007) Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity 26(4):503–517. doi:10.1016/j.immuni.2007.03.006
Mamessier E, Sylvain A, Thibult ML, Houvenaeghel G, Jacquemier J, Castellano R, Goncalves A, Andre P, Romagne F, Thibault G, Viens P, Birnbaum D, Bertucci F, Moretta A, Olive D (2011) Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. J Clin Investig 121(9):3609–3622. doi:10.1172/JCI45816
Martinet L, Smyth MJ (2015) Balancing natural killer cell activation through paired receptors. Nat Rev Immunol 15(4):243–254. doi:10.1038/nri3799
Martin-Fontecha A, Thomsen LL, Brett S, Gerard C, Lipp M, Lanzavecchia A, Sallusto F (2004) Induced recruitment of NK cells to lymph nodes provides IFN-gamma for T(H)1 priming. Nat Immunol 5(12):1260–1265. doi:10.1038/ni1138
McGilvray RW, Eagle RA, Watson NF, Al-Attar A, Ball G, Jafferji I, Trowsdale J, Durrant LG (2009) NKG2D ligand expression in human colorectal cancer reveals associations with prognosis and evidence for immunoediting. Clinical Cancer Res Official J Am Assoc Cancer Res 15(22):6993–7002. doi:10.1158/1078-0432.CCR-09-0991
McIntyre KW, Welsh RM (1986) Accumulation of natural killer and cytotoxic T large granular lymphocytes in the liver during virus infection. J Exp Med 164(5):1667–1681
Mittal D, Young A, Stannard K, Yong M, Teng MW, Allard B, Stagg J, Smyth MJ (2014) Antimetastatic effects of blocking PD-1 and the adenosine A2A receptor. Cancer Res 74(14):3652–3658. doi:10.1158/0008-5472.CAN-14-0957
Mizutani T, Neugebauer N, Putz EM, Moritz N, Simma O, Zebedin-Brandl E, Gotthardt D, Warsch W, Eckelhart E, Kantner HP, Kalinke U, Lienenklaus S, Weiss S, Strobl B, Muller M, Sexl V, Stoiber D (2012) Conditional IFNAR1 ablation reveals distinct requirements of Type I IFN signaling for NK cell maturation and tumor surveillance. Oncoimmunology 1(7):1027–1037. doi:10.4161/onci.21284
Mocikat R, Braumuller H, Gumy A, Egeter O, Ziegler H, Reusch U, Bubeck A, Louis J, Mailhammer R, Riethmuller G, Koszinowski U, Rocken M (2003) Natural killer cells activated by MHC class I(low) targets prime dendritic cells to induce protective CD8 T cell responses. Immunity 19(4):561–569
Morandi B, Mortara L, Chiossone L, Accolla RS, Mingari MC, Moretta L, Moretta A, Ferlazzo G (2012) Dendritic cell editing by activated natural killer cells results in a more protective cancer-specific immune response. PLoS ONE 7(6):e39170. doi:10.1371/journal.pone.0039170
Moretta A, Marcenaro E, Sivori S, Della Chiesa M, Vitale M, Moretta L (2005) Early liaisons between cells of the innate immune system in inflamed peripheral tissues. Trends Immunol 26(12):668–675. doi:10.1016/j.it.2005.09.008
Newman KC, Riley EM (2007) Whatever turns you on: accessory-cell-dependent activation of NK cells by pathogens. Nat Rev Immunol 7(4):279–291. doi:10.1038/nri2057
Orange JS (2013) Natural killer cell deficiency. J Allergy Clinical Immunol 132(3):515–525; quiz 526. doi:10.1016/j.jaci.2013.07.020
Oshimi Y, Oda S, Honda Y, Nagata S, Miyazaki S (1996) Involvement of Fas ligand and Fas-mediated pathway in the cytotoxicity of human natural killer cells. J Immunol 157(7):2909–2915
O’Sullivan T, Saddawi-Konefka R, Vermi W, Koebel CM, Arthur C, White JM, Uppaluri R, Andrews DM, Ngiow SF, Teng MW, Smyth MJ, Schreiber RD, Bui JD (2012) Cancer immunoediting by the innate immune system in the absence of adaptive immunity. J Exp Med 209(10):1869–1882. doi:10.1084/jem.20112738
Pachynski RK, Zabel BA, Kohrt HE, Tejeda NM, Monnier J, Swanson CD, Holzer AK, Gentles AJ, Sperinde GV, Edalati A, Hadeiba HA, Alizadeh AA, Butcher EC (2012) The chemoattractant chemerin suppresses melanoma by recruiting natural killer cell antitumor defenses. J Exp Med 209(8):1427–1435. doi:10.1084/jem.20112124
Pahl JH, Ruslan SE, Buddingh EP, Santos SJ, Szuhai K, Serra M, Gelderblom H, Hogendoorn PC, Egeler RM, Schilham MW, Lankester AC (2012) Anti-EGFR antibody cetuximab enhances the cytolytic activity of natural killer cells toward osteosarcoma. Clinical Cancer Res Official J Am Assoc Cancer Res 18(2):432–441. doi:10.1158/1078-0432.CCR-11-2277
Pan PY, Gu P, Li Q, Xu D, Weber K, Chen SH (2004) Regulation of dendritic cell function by NK cells: mechanisms underlying the synergism in the combination therapy of IL-12 and 4-1BB activation. J Immunol 172(8):4779–4789
Pardo J, Balkow S, Anel A, Simon MM (2002) Granzymes are essential for natural killer cell-mediated and perf-facilitated tumor control. Eur J Immunol 32(10):2881–2887. doi:10.1002/1521-4141(2002010)32:10<2881:AID-IMMU2881>3.0.CO;2-K
Pardoll DM (2012) The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 12(4):252–264. doi:10.1038/nrc3239
Parham P (2005) MHC class I molecules and KIRs in human history, health and survival. Nat Rev Immunol 5(3):201–214. doi:10.1038/nri1570
Parolini S, Santoro A, Marcenaro E, Luini W, Massardi L, Facchetti F, Communi D, Parmentier M, Majorana A, Sironi M, Tabellini G, Moretta A, Sozzani S (2007) The role of chemerin in the colocalization of NK and dendritic cell subsets into inflamed tissues. Blood 109(9):3625–3632. doi:10.1182/blood-2006-08-038844
Pende D, Cantoni C, Rivera P, Vitale M, Castriconi R, Marcenaro S, Nanni M, Biassoni R, Bottino C, Moretta A, Moretta L (2001) Role of NKG2D in tumor cell lysis mediated by human NK cells: cooperation with natural cytotoxicity receptors and capability of recognizing tumors of nonepithelial origin. Eur J Immunol 31(4):1076–1086. doi:10.1002/1521-4141(200104)31:4<1076:AID-IMMU1076>3.0.CO;2-Y
Pende D, Spaggiari GM, Marcenaro S, Martini S, Rivera P, Capobianco A, Falco M, Lanino E, Pierri I, Zambello R, Bacigalupo A, Mingari MC, Moretta A, Moretta L (2005) Analysis of the receptor-ligand interactions in the natural killer-mediated lysis of freshly isolated myeloid or lymphoblastic leukemias: evidence for the involvement of the Poliovirus receptor (CD155) and Nectin-2 (CD112). Blood 105(5):2066–2073. doi:10.1182/blood-2004-09-3548
Platonova S, Cherfils-Vicini J, Damotte D, Crozet L, Vieillard V, Validire P, Andre P, Dieu-Nosjean MC, Alifano M, Regnard JF, Fridman WH, Sautes-Fridman C, Cremer I (2011) Profound coordinated alterations of intratumoral NK cell phenotype and function in lung carcinoma. Cancer Res 71(16):5412–5422. doi:10.1158/0008-5472.CAN-10-4179
Raulet DH (2003) Roles of the NKG2D immunoreceptor and its ligands. Nat Rev Immunol 3(10):781–790. doi:10.1038/nri1199
Raulet DH, Vance RE (2006) Self-tolerance of natural killer cells. Nat Rev Immunol 6(7):520–531. doi:10.1038/nri1863
Remark R, Alifano M, Cremer I, Lupo A, Dieu-Nosjean MC, Riquet M, Crozet L, Ouakrim H, Goc J, Cazes A, Flejou JF, Gibault L, Verkarre V, Regnard JF, Pages ON, Oudard S, Mlecnik B, Sautes-Fridman C, Fridman WH, Damotte D (2013) Characteristics and clinical impacts of the immune environments in colorectal and renal cell carcinoma lung metastases: influence of tumor origin. Clinical Cancer Res Official J Am Assoc Cancer Res 19(15):4079–4091. doi:10.1158/1078-0432.CCR-12-3847
Romagnani C, Babic M (2014) NK/DC crosstalk in immunosurveillance: a broken relationship caused by WASP-deficiency. Eur J Immunol 44(4):958–961. doi:10.1002/eji.201444514
Santoni A, Herberman RB, Holden HT (1979) Correlation between natural and antibody-dependent cell-mediated cytotoxicity against tumor targets in the mouse. II. Characterization of the effector cells. J Natl Cancer Inst 63(4):995–1003
Sarkar S, Germeraad WT, Rouschop KM, Steeghs EM, van Gelder M, Bos GM, Wieten L (2013) Hypoxia induced impairment of NK cell cytotoxicity against multiple myeloma can be overcome by IL-2 activation of the NK cells. PLoS ONE 8(5):e64835. doi:10.1371/journal.pone.0064835
Sathe P, Delconte RB, Souza-Fonseca-Guimaraes F, Seillet C, Chopin M, Vandenberg CJ, Rankin LC, Mielke LA, Vikstrom I, Kolesnik TB, Nicholson SE, Vivier E, Smyth MJ, Nutt SL, Glaser SP, Strasser A, Belz GT, Carotta S, Huntington ND (2014) Innate immunodeficiency following genetic ablation of Mcl1 in natural killer cells. Nature Commun 5:4539. doi:10.1038/ncomms5539
Satoh-Takayama N, Vosshenrich CA, Lesjean-Pottier S, Sawa S, Lochner M, Rattis F, Mention JJ, Thiam K, Cerf-Bensussan N, Mandelboim O, Eberl G, Di Santo JP (2008) Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity 29(6):958–970. doi:10.1016/j.immuni.2008.11.001
Sceneay J, Chow MT, Chen A, Halse HM, Wong CS, Andrews DM, Sloan EK, Parker BS, Bowtell DD, Smyth MJ, Moller A (2012) Primary tumor hypoxia recruits CD11b+/Ly6Cmed/Ly6G+ immune suppressor cells and compromises NK cell cytotoxicity in the premetastatic niche. Cancer Res 72(16):3906–3911. doi:10.1158/0008-5472.CAN-11-3873
Sconocchia G, Arriga R, Tornillo L, Terracciano L, Ferrone S, Spagnoli GC (2012) Melanoma cells inhibit NK cell functions. Cancer Res 72 (20):5428–5429; author reply 5430. doi:10.1158/0008-5472.CAN-12-1181
Screpanti V, Wallin RP, Ljunggren HG, Grandien A (2001) A central role for death receptor-mediated apoptosis in the rejection of tumors by NK cells. J Immunol 167(4):2068–2073
Seaman WE, Sleisenger M, Eriksson E, Koo GC (1987) Depletion of natural killer cells in mice by monoclonal antibody to NK-1.1. Reduction in host defense against malignancy without loss of cellular or humoral immunity. J Immunol 138(12):4539–4544
Seillet C, Huntington ND, Gangatirkar P, Axelsson E, Minnich M, Brady HJ, Busslinger M, Smyth MJ, Belz GT, Carotta S (2014) Differential requirement for Nfil3 during NK cell development. J Immunol 192(6):2667–2676. doi:10.4049/jimmunol.1302605
Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, Schreiber RD (2001) IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 410(6832):1107–1111. doi:10.1038/35074122
Shimizu K, Asakura M, Fujii S (2011) Prolonged antitumor NK cell reactivity elicited by CXCL10-expressing dendritic cells licensed by CD40L+ CD4+ memory T cells. J Immunol 186(10):5927–5937. doi:10.4049/jimmunol.1003351
Shresta S, MacIvor DM, Heusel JW, Russell JH, Ley TJ (1995) Natural killer and lymphokine-activated killer cells require granzyme B for the rapid induction of apoptosis in susceptible target cells. Proc Natl Acad Sci USA 92(12):5679–5683
Sivori S, Pende D, Bottino C, Marcenaro E, Pessino A, Biassoni R, Moretta L, Moretta A (1999) NKp46 is the major triggering receptor involved in the natural cytotoxicity of fresh or cultured human NK cells. Correlation between surface density of NKp46 and natural cytotoxicity against autologous, allogeneic or xenogeneic target cells. Eur J Immunol 29(5):1656–1666. doi:10.1002/(SICI)1521-4141(199905)29:05<1656:AID-IMMU1656>3.0.CO;2-1
Sivori S, Parolini S, Falco M, Marcenaro E, Biassoni R, Bottino C, Moretta L, Moretta A (2000) 2B4 functions as a co-receptor in human NK cell activation. Eur J Immunol 30(3):787–793. doi:10.1002/1521-4141(200003)30:3<787:AID-IMMU787>3.0.CO;2-I
Sivori S, Falco M, Marcenaro E, Parolini S, Biassoni R, Bottino C, Moretta L, Moretta A (2002) Early expression of triggering receptors and regulatory role of 2B4 in human natural killer cell precursors undergoing in vitro differentiation. Proc Natl Acad Sci USA 99(7):4526–4531. doi:10.1073/pnas.072065999
Sivori S, Falco M, Della Chiesa M, Carlomagno S, Vitale M, Moretta L, Moretta A (2004) CpG and double-stranded RNA trigger human NK cells by Toll-like receptors: induction of cytokine release and cytotoxicity against tumors and dendritic cells. Proc Natl Acad Sci USA 101(27):10116–10121. doi:10.1073/pnas.0403744101
Smyth MJ, Johnstone RW (2000) Role of TNF in lymphocyte-mediated cytotoxicity. Microsc Res Tech 50(3):196–208. doi:10.1002/1097-0029(20000801)50:3<196:AID-JEMT3>3.0.CO;2-9
Smyth MJ, Kelly JM, Baxter AG, Korner H, Sedgwick JD (1998) An essential role for tumor necrosis factor in natural killer cell-mediated tumor rejection in the peritoneum. J Exp Med 188(9):1611–1619
Smyth MJ, Thia KY, Cretney E, Kelly JM, Snook MB, Forbes CA, Scalzo AA (1999) Perforin is a major contributor to NK cell control of tumor metastasis. J Immunol 162(11):6658–6662
Smyth MJ, Taniguchi M, Street SE (2000a) The anti-tumor activity of IL-12: mechanisms of innate immunity that are model and dose dependent. J Immunol 165(5):2665–2670
Smyth MJ, Thia KY, Street SE, MacGregor D, Godfrey DI, Trapani JA (2000b) Perforin-mediated cytotoxicity is critical for surveillance of spontaneous lymphoma. J Exp Med 192(5):755–760
Smyth MJ, Crowe NY, Godfrey DI (2001) NK cells and NKT cells collaborate in host protection from methylcholanthrene-induced fibrosarcoma. Int Immunol 13(4):459–463
Smyth MJ, Hayakawa Y, Takeda K, Yagita H (2002) New aspects of natural-killer-cell surveillance and therapy of cancer. Nat Rev Cancer 2(11):850–861. doi:10.1038/nrc928
Smyth MJ, Swann J, Kelly JM, Cretney E, Yokoyama WM, Diefenbach A, Sayers TJ, Hayakawa Y (2004) NKG2D recognition and perforin effector function mediate effective cytokine immunotherapy of cancer. J Exp Med 200(10):1325–1335. doi:10.1084/jem.20041522
Smyth MJ, Swann J, Cretney E, Zerafa N, Yokoyama WM, Hayakawa Y (2005) NKG2D function protects the host from tumor initiation. J Exp Med 202(5):583–588. doi:10.1084/jem.20050994
Sojka DK, Plougastel-Douglas B, Yang L, Pak-Wittel MA, Artyomov MN, Ivanova Y, Zhong C, Chase JM, Rothman PB, Yu J, Riley JK, Zhu J, Tian Z, Yokoyama WM (2014) Tissue-resident natural killer (NK) cells are cell lineages distinct from thymic and conventional splenic NK cells. eLife 3:e01659. doi:10.7554/eLife.01659
Soriani A, Zingoni A, Cerboni C, Iannitto ML, Ricciardi MR, Di Gialleonardo V, Cippitelli M, Fionda C, Petrucci MT, Guarini A, Foa R, Santoni A (2009) ATM-ATR-dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood 113(15):3503–3511. doi:10.1182/blood-2008-08-173914
Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie AN, Mebius RE, Powrie F, Vivier E (2013) Innate lymphoid cells–a proposal for uniform nomenclature. Nat Rev Immunol 13(2):145–149. doi:10.1038/nri3365
Srivastava RM, Lee SC, Andrade Filho PA, Lord CA, Jie HB, Davidson HC, Lopez-Albaitero A, Gibson SP, Gooding WE, Ferrone S, Ferris RL (2013) Cetuximab-activated natural killer and dendritic cells collaborate to trigger tumor antigen-specific T-cell immunity in head and neck cancer patients. Clinical Cancer Res Official J Am Assoc Cancer Res 19(7):1858–1872. doi:10.1158/1078-0432.CCR-12-2426
Strbo N, Oizumi S, Sotosek-Tokmadzic V, Podack ER (2003) Perforin is required for innate and adaptive immunity induced by heat shock protein gp96. Immunity 18(3):381–390
Street SE, Cretney E, Smyth MJ (2001) Perforin and interferon-gamma activities independently control tumor initiation, growth, and metastasis. Blood 97(1):192–197
Street SE, Trapani JA, MacGregor D, Smyth MJ (2002) Suppression of lymphoma and epithelial malignancies effected by interferon gamma. J Exp Med 196(1):129–134
Tahara-Hanaoka S, Shibuya K, Kai H, Miyamoto A, Morikawa Y, Ohkochi N, Honda S, Shibuya A (2006) Tumor rejection by the poliovirus receptor family ligands of the DNAM-1 (CD226) receptor. Blood 107(4):1491–1496. doi:10.1182/blood-2005-04-1684
Takeda K, Hayakawa Y, Smyth MJ, Kayagaki N, Yamaguchi N, Kakuta S, Iwakura Y, Yagita H, Okumura K (2001) Involvement of tumor necrosis factor-related apoptosis-inducing ligand in surveillance of tumor metastasis by liver natural killer cells. Nat Med 7(1):94–100. doi:10.1038/83416
Takeda K, Smyth MJ, Cretney E, Hayakawa Y, Kayagaki N, Yagita H, Okumura K (2002) Critical role for tumor necrosis factor-related apoptosis-inducing ligand in immune surveillance against tumor development. J Exp Med 195(2):161–169
Takeda K, Cretney E, Hayakawa Y, Ota T, Akiba H, Ogasawara K, Yagita H, Kinoshita K, Okumura K, Smyth MJ (2005) TRAIL identifies immature natural killer cells in newborn mice and adult mouse liver. Blood 105(5):2082–2089. doi:10.1182/blood-2004-08-3262
Talmadge JE, Meyers KM, Prieur DJ, Starkey JR (1980) Role of NK cells in tumour growth and metastasis in beige mice. Nature 284(5757):622–624
Terme M, Ullrich E, Aymeric L, Meinhardt K, Coudert JD, Desbois M, Ghiringhelli F, Viaud S, Ryffel B, Yagita H, Chen L, Mecheri S, Kaplanski G, Prevost-Blondel A, Kato M, Schultze JL, Tartour E, Kroemer G, Degli-Esposti M, Chaput N, Zitvogel L (2012) Cancer-induced immunosuppression: IL-18-elicited immunoablative NK cells. Cancer Res 72(11):2757–2767. doi:10.1158/0008-5472.CAN-11-3379
Tu MM, Mahmoud AB, Wight A, Mottashed A, Belanger S, Rahim MM, Abou-Samra E, Makrigiannis AP (2014) Ly49 family receptors are required for cancer immunosurveillance mediated by natural killer cells. Cancer Res 74(14):3684–3694. doi:10.1158/0008-5472.CAN-13-3021
Urosevic M, Dummer R (2008) Human leukocyte antigen-G and cancer immunoediting. Cancer Res 68(3):627–630. doi:10.1158/0008-5472.CAN-07-2704
Vaidya SV, Stepp SE, McNerney ME, Lee JK, Bennett M, Lee KM, Stewart CL, Kumar V, Mathew PA (2005) Targeted disruption of the 2B4 gene in mice reveals an in vivo role of 2B4 (CD244) in the rejection of B16 melanoma cells. J Immunol 174(2):800–807
van den Broek MF, Kagi D, Zinkernagel RM, Hengartner H (1995) Perforin dependence of natural killer cell-mediated tumor control in vivo. Eur J Immunol 25(12):3514–3516. doi:10.1002/eji.1830251246
van den Broek ME, Kagi D, Ossendorp F, Toes R, Vamvakas S, Lutz WK, Melief CJ, Zinkernagel RM, Hengartner H (1996) Decreased tumor surveillance in perforin-deficient mice. J Exp Med 184(5):1781–1790
Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ (2011) Natural innate and adaptive immunity to cancer. Annu Rev Immunol 29:235–271. doi:10.1146/annurev-immunol-031210-101324
Vitale M, Bottino C, Sivori S, Sanseverino L, Castriconi R, Marcenaro E, Augugliaro R, Moretta L, Moretta A (1998) NKp44, a novel triggering surface molecule specifically expressed by activated natural killer cells, is involved in non-major histocompatibility complex-restricted tumor cell lysis. J Exp Med 187(12):2065–2072
Vitale M, Cantoni C, Pietra G, Mingari MC, Moretta L (2014) Effect of tumor cells and tumor microenvironment on NK-cell function. Eur J Immunol 44(6):1582–1592. doi:10.1002/eji.201344272
Vivier E (2006) What is natural in natural killer cells? Immunol Lett 107(1):1–7. doi:10.1016/j.imlet.2006.07.004
Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S (2008) Functions of natural killer cells. Nat Immunol 9(5):503–510. doi:10.1038/ni1582
Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, Yokoyama WM, Ugolini S (2011) Innate or adaptive immunity? The example of natural killer cells. Science 331(6013):44–49. doi:10.1126/science.1198687
Voskoboinik I, Smyth MJ, Trapani JA (2006) Perforin-mediated target-cell death and immune homeostasis. Nat Rev Immunol 6(12):940–952. doi:10.1038/nri1983
Walzer T, Blery M, Chaix J, Fuseri N, Chasson L, Robbins SH, Jaeger S, Andre P, Gauthier L, Daniel L, Chemin K, Morel Y, Dalod M, Imbert J, Pierres M, Moretta A, Romagne F, Vivier E (2007) Identification, activation, and selective in vivo ablation of mouse NK cells via NKp46. Proc Natl Acad Sci USA 104(9):3384–3389. doi:10.1073/pnas.0609692104
Weiner LM, Surana R, Wang S (2010) Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nat Rev Immunol 10(5):317–327. doi:10.1038/nri2744
Wendel M, Galani IE, Suri-Payer E, Cerwenka A (2008) Natural killer cell accumulation in tumors is dependent on IFN-gamma and CXCR3 ligands. Cancer Res 68(20):8437–8445. doi:10.1158/0008-5472.CAN-08-1440
Wilk E, Kalippke K, Buyny S, Schmidt RE, Jacobs R (2008) New aspects of NK cell subset identification and inference of NK cells’ regulatory capacity by assessing functional and genomic profiles. Immunobiology 213(3–4):271–283. doi:10.1016/j.imbio.2007.10.012
Yajima T, Nishimura H, Wajjwalku W, Harada M, Kuwano H, Yoshikai Y (2002) Overexpression of interleukin-15 in vivo enhances antitumor activity against MHC class I-negative and -positive malignant melanoma through augmented NK activity and cytotoxic T-cell response. Int J Cancer 99(4):573–578. doi:10.1002/ijc.10395
Young A, Mittal D, Stagg J, Smyth MJ (2014) Targeting cancer-derived adenosine: new therapeutic approaches. Cancer Discov 4(8):879–888. doi:10.1158/2159-8290.CD-14-0341
Zamai L, Ahmad M, Bennett IM, Azzoni L, Alnemri ES, Perussia B (1998) Natural killer (NK) cell-mediated cytotoxicity: differential use of TRAIL and Fas ligand by immature and mature primary human NK cells. J Exp Med 188(12):2375–2380
Zhang C, Zhang J, Tian Z (2006) The regulatory effect of natural killer cells: do “NK-reg cells” exist? Cell Mol Immunol 3(4):241–254
Zhao BG, Vasilakos JP, Tross D, Smirnov D, Klinman DM (2014) Combination therapy targeting toll like receptors 7, 8 and 9 eliminates large established tumors. J Immunother Cancer 2:12. doi:10.1186/2051-1426-2-12
Zheng LM, Ojcius DM, Garaud F, Roth C, Maxwell E, Li Z, Rong H, Chen J, Wang XY, Catino JJ, King I (1996) Interleukin-10 inhibits tumor metastasis through an NK cell-dependent mechanism. J Exp Med 184(2):579–584
Acknowledgments
M.J.S. is supported by a NH&MRC Australia Fellowship (628623) and Program Grant (1013667).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Guillerey, C., Smyth, M.J. (2015). NK Cells and Cancer Immunoediting. In: Vivier, E., Di Santo, J., Moretta, A. (eds) Natural Killer Cells. Current Topics in Microbiology and Immunology, vol 395. Springer, Cham. https://doi.org/10.1007/82_2015_446
Download citation
DOI: https://doi.org/10.1007/82_2015_446
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-23915-6
Online ISBN: 978-3-319-23916-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)